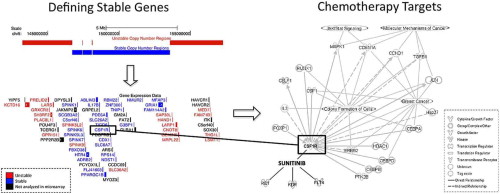
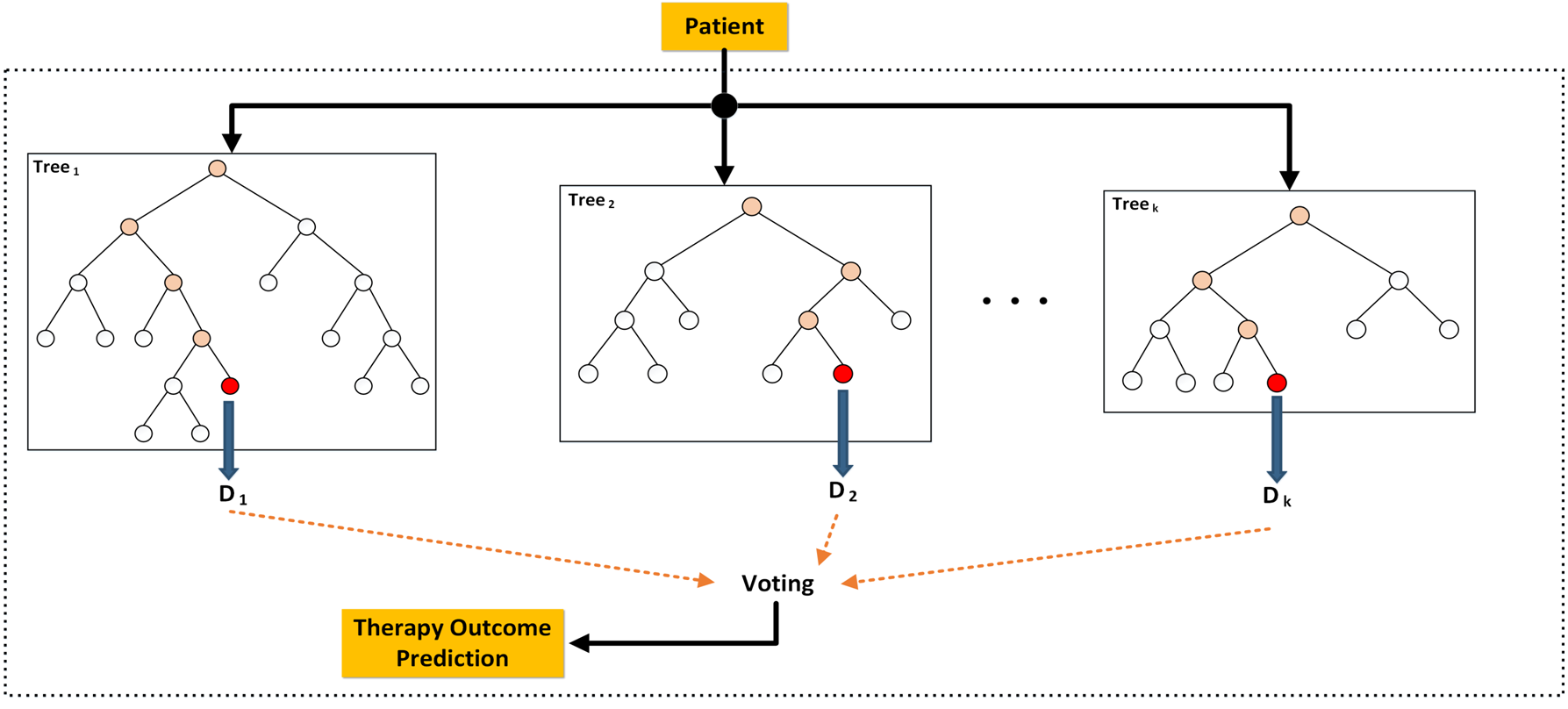
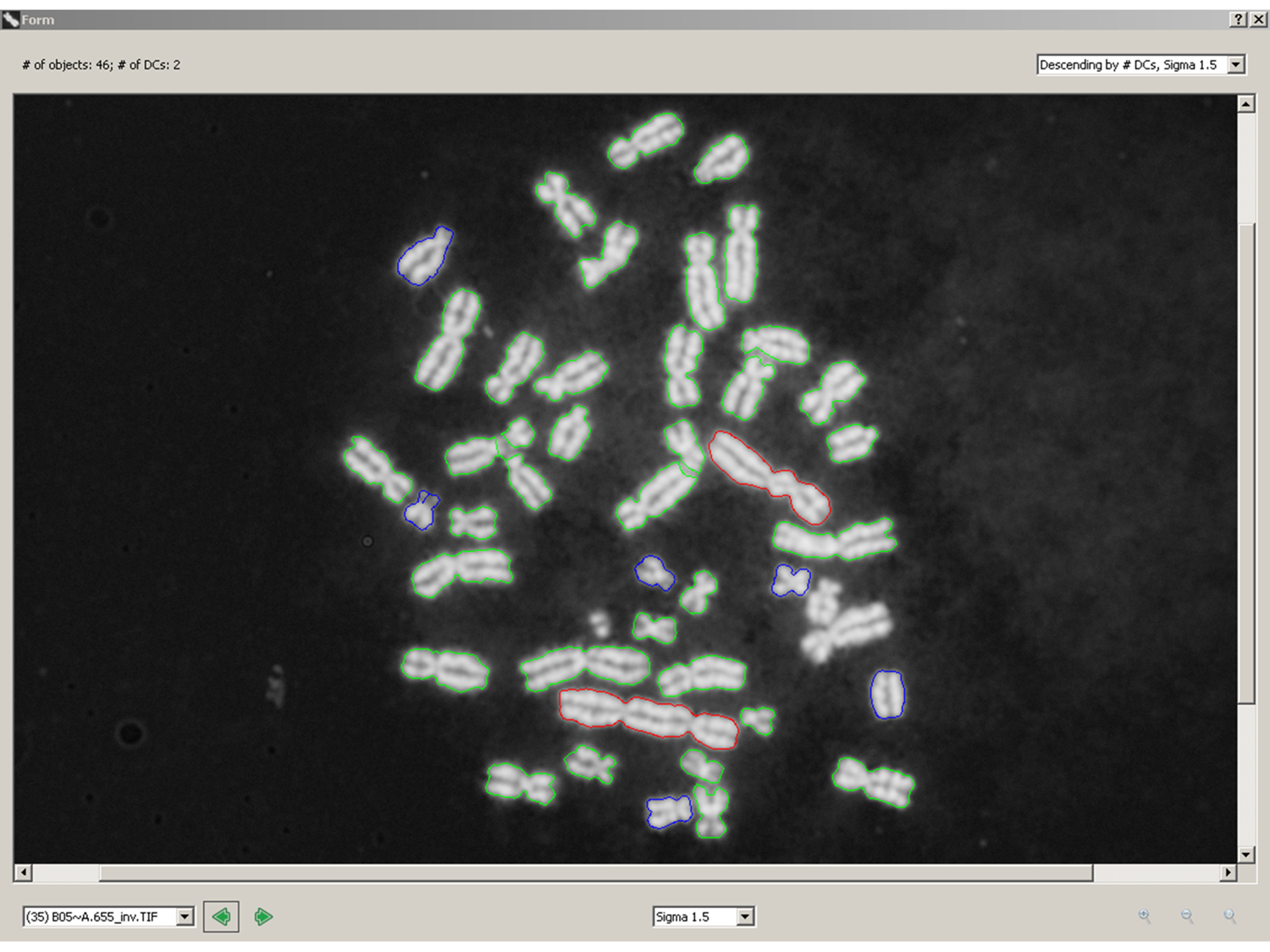
Citation:
Lu R, Mucaki E and Rogan PK. Discovery and Validation of Information Theory-Based Transcription Factor and Cofactor Binding Site Motifs, Nucleic Acids Research. DOI: 10.1093/nar/gkw1036 (pdf)
Download:
Copyright licence (CC-BY)
Manuscript with Figures – Lu, Mucaki and Rogan, Nucl. Acids Res. 2016
Supplementary – Table S1; Table S2; Table S3; Table S4; Table S5; Table S6; Table S7; Table S8