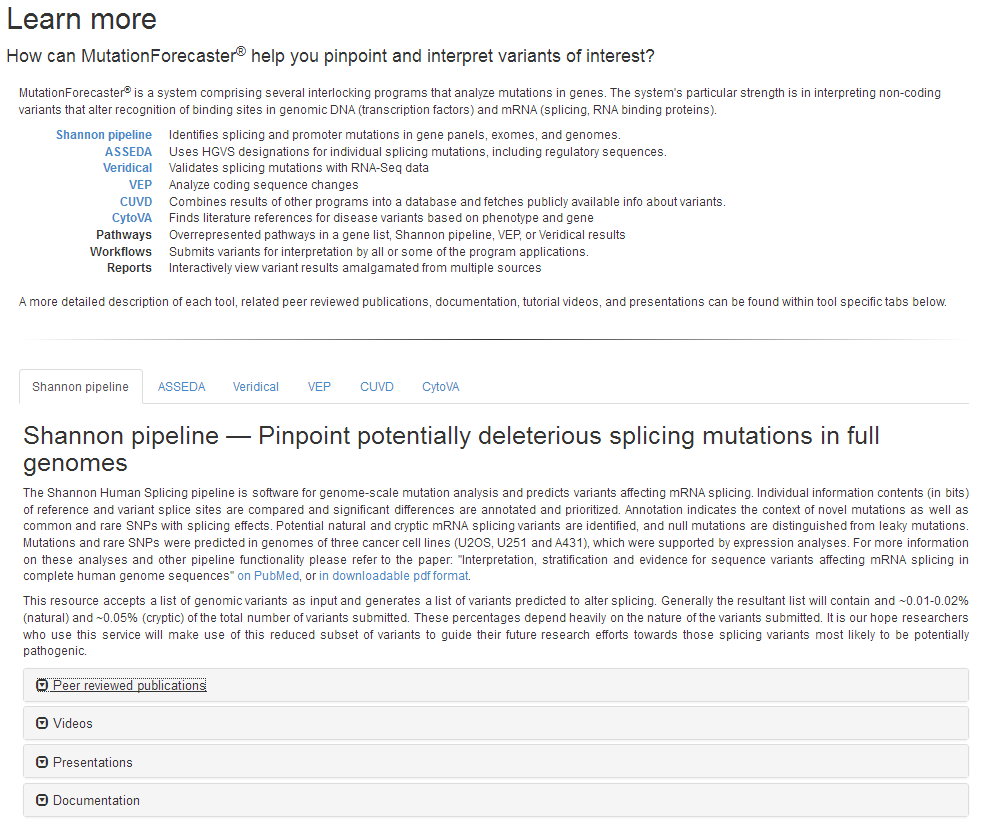
We have restructured and added new content to the MutationForecaster® “Learn More” page:

Yang et al. Prevalence and spectrum of germline rare variants in BRCA1/2 and PALB2 among breast cancer cases in Sarawak, Malaysia. Breast Cancer Research and Treatment, 2017. (Link to paper [pdf])
This study applies information theory based mutation analysis to interpret pathogenic variants and variants of uncertain significance in these genes.

Peter Rogan will be presenting “A Unified Framework For Prioritization Of Variants Of Uncertain Significance In Hereditary Breast And Ovarian Cancer” at Variant Detection 2017 in Santiago de Compostela Spain on June 5, 2017.
Coauthors are Eliseos Mucaki1, Natasha Caminsky1, Ruipeng Lu1, Joan Knoll1,2 and Peter Rogan1,2. 1University of Western Ontario, 2CytoGnomix Inc.
Abstract:
Purpose: A significant proportion of HBOC patients receive uninformative genetic testing results, an issue exacerbated by the overwhelming quantity of variants of uncertain significance identified. We apply information theory (IT) to predict and analyze non-coding variants of uncertain significance (VUS) in regulatory, coding, and intronic regions based on changes in binding sites in these genes. This provides a unifying framework where, aside from protein coding changes, pathogenic variants occurring within sequence elements can be prioritized based 19 on their recognition by proteins involved in mRNA splicing, transcription, and untranslated region binding and structure. To support the utilization of IT analysis, we established IT-based variant interpretation accuracy by performing a comprehensive review of mutations altering mRNA splicing in rare and common diseases1.
Methods: We captured and enriched for coding and non-coding variants in genes known or suspected to increase HBOC risk. Custom oligonucleotide baits spanning the complete coding, non-coding, and intergenic regions 10 kb up- and downstream of ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, TP53, ATM, BARD1, BRCA1, BRCA2, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51B, STK11, TP53, and XRCC2 were synthesized for solution hybridization enrichment. Aside from protein coding and copy number changes, IT-based sequence analysis was used to identify and prioritize pathogenic non-coding variants that occurred within sequence elements predicted to be recognized by proteins or protein complexes involved in mRNA splicing, transcription, and untranslated region (UTR) binding and structure. Mutation-associated affinity changes were computed in transcription factor (TFBSs)2, splicing regulatory (SRBSs)3, and RNA-binding protein (RBBSs)4 binding sites following mutation. This approach was supplemented by in silico and laboratory analysis of UTR structure.
Results: Unique and divergent repetitive sequences were sequenced in 379 high-risk, patients without identified mutations in BRCA1/2. We identified 47,501 unique variants and we prioritized 429 variants. The methods were first applied in 7 complete genes (ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, TP53) in 102 anonymized individuals (15,311 variants)4, then validated in 287 patients in an ethics board approved study (38,372 variants)5. In the validation study, we prioritized variants affecting the strengths of 10 splice sites (4 natural, 6 cryptic), 148 SRBS, 36 TFBS, and 31 RBBS. Three variants were also prioritized based on their predicted effects on mRNA secondary (2°) structure, and 17 for pseudoexon activation. Additionally, 4 frameshift, 2 in-frame deletions, and 5 stopgain mutations were identified. Multifactorial cosegregation analysis further reduced the set of candidate pathogenic variants in some families.
Conclusion: Complete gene sequence analysis followed by a unified framework can be used to interpret non-coding variants that may affect gene expression. When combined with pedigree information, complete gene sequence analysis can distill large numbers of VUS among a wide spectrum of functional mutation types to a limited set of variants for downstream functional and co-segregation analysis. References: 1Caminsky et al. F1000Res 3:282, 2015; 2Lu et al. Nucleic Acids Res. doi: 10.1093/nar/gkw1036, 2016; 3Mucaki et al. Hum. Mut. 34:557-565. 2013; 4Mucaki et al. BMC Med Genomics. 9:19, 2016; 5Caminsky et al. Hum. Mut. 37:640-52, 2016.
CytoGnomix has delivered the Automated Dicentric Chromosome Identifier and Dose Estimator system to the Healthy Environments and Consumer Safety Branch at Health Canada, Government of Canada and to the Biodosimetry laboratory at Canadian Nuclear Laboratories. Each lab received two licensed versions of the product on a state of the art MSI portable computer and two full days of training on the systems. Experienced personnel will use ADCI to perform tests on radiation exposed blood samples from international comparative biodosimetry exercises. They will evaluate accuracy of dose estimation and speed of analysis.

Our article:
“Expedited radiation biodosimetry by automated dicentric chromosome identification and dose estimation”
Ben Shirley 1^ , Yanxin Li 1^ , Joan H. M. Knoll 1,2 , and Peter K. Rogan 1,3
1 CytoGnomix, Departments of 2 Pathology and Laboratory Medicine and 3 Biochemistry, Western
University, London, ON Canada
has been accepted for publication in the Journal of Visualized Experiments. The paper describes, in detail, the protocol for use of the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI). It will be accompanied by a professionally produced video that illustrates how the software is used to determine radiation dose from metaphase cell images.
Short abstract:
The cytogenetic dicentric chromosome (DC) assay quantifies exposure to ionizing radiation. The Automated Dicentric Chromosome Identifier and Dose Estimator software accurately and rapidly estimates biological dose from DCs in metaphase cells. It distinguishes monocentric chromosomes and other objects from DCs, and estimates biological radiation dose from the frequency of DCs.
![]() CytoGnomix will be exhibiting at the 22nd Nuclear Medical Defense Conference, next week (May 8th to 11th 2017) in Munich, Germany. We will be introducing our biodosimetry product, the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) at the meeting. We will also be presenting a poster on novel, patent pending methods to automatically curate metaphase cell selection and chromosomes in digital images.
CytoGnomix will be exhibiting at the 22nd Nuclear Medical Defense Conference, next week (May 8th to 11th 2017) in Munich, Germany. We will be introducing our biodosimetry product, the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) at the meeting. We will also be presenting a poster on novel, patent pending methods to automatically curate metaphase cell selection and chromosomes in digital images.
US Patent 9,624,549 has been issued!
“Stable gene targets in breast cancer and use thereof for optimizing therapy. ” Peter K Rogan and Joan Knoll. (link)
This technology is the basis of biochemically inspired chemotherapy prediction by machine learning (http://chemotherapy.cytognomix.com).
Rogan PK, Mucaki EJ, Baranova K, Dorman S, Knoll JHM. Predicting responses to chemotherapies by biochemically-inspired machine learning. Innovative Approaches to Optimal Cancer Care in Canada, Canadian Partnership against Cancer, Toronto, Apr. 6-8, 2017. (Link to abstract)
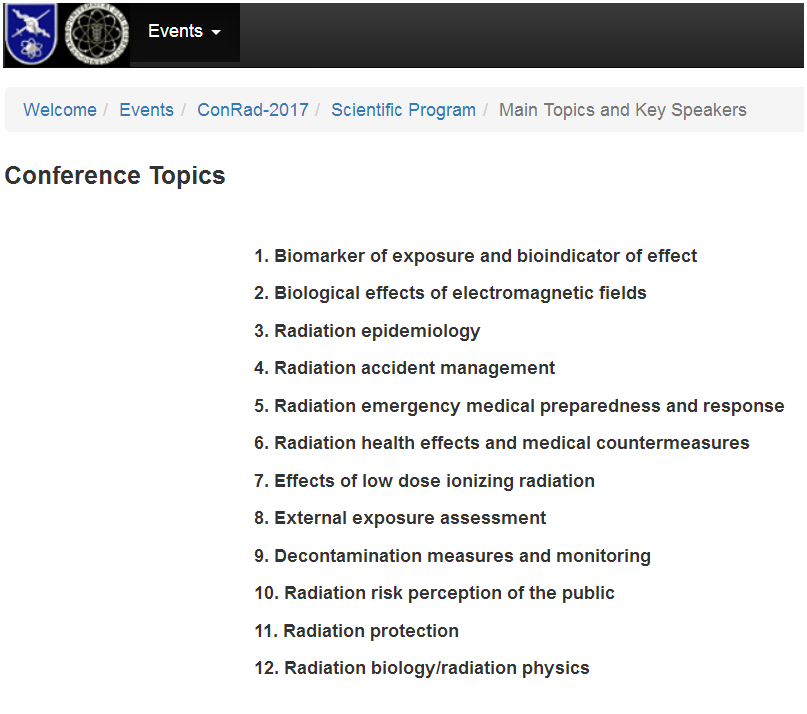
Accurate Cytogenetic Biodosimetry Through Automation Of Dicentric Chromosome Curation And Metaphase Cell Selection
CytoGnomix has finalized our contract with Public Works Government Services Canada under the Build in Canada Innovation Program. This agreement licenses the Automated Dicentric Chromosome Identifier (ADCI) to the Consumer and Clinical Radiation Protection Bureau at Health Canada and Canadian Nuclear Laboratories and provides on-site training to these labs. These biodosimetry reference labs will test the software and provide feedback. Test results will support CytoGnomix’s submitted application to the Medical Device Bureau at Health Canada.
We have published a new version of:
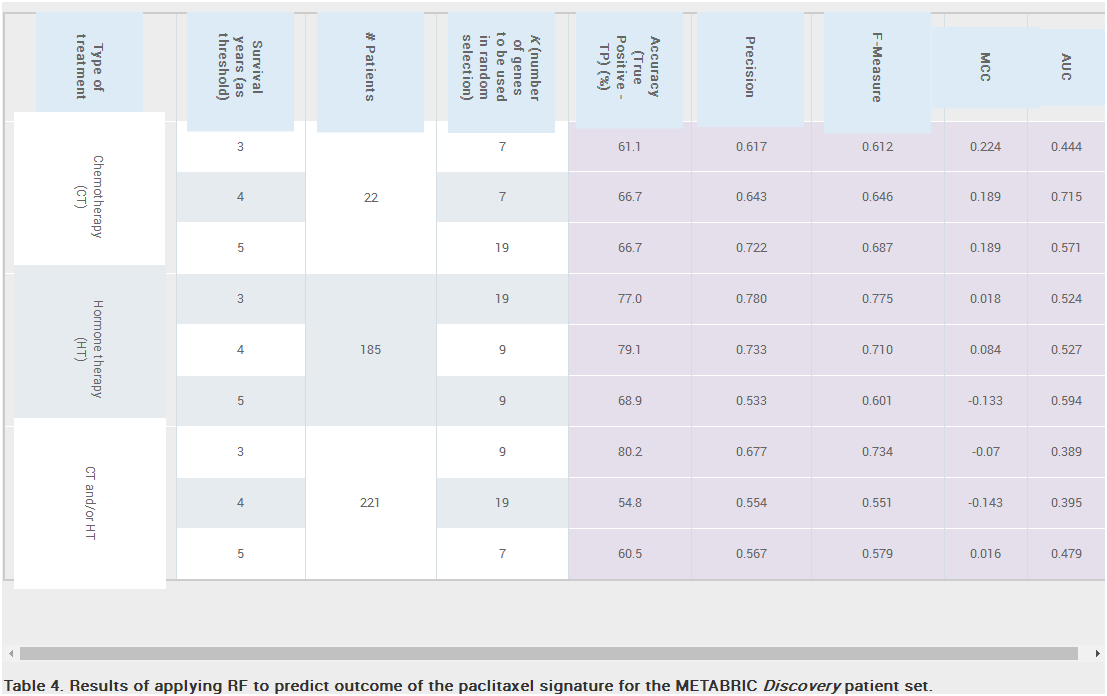
Predicting Outcomes of Hormone and Chemotherapy in the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) Study by Biochemically-inspired Machine Learning. F1000Research 2017, 5:2124 (doi:10.12688/f1000research.9417.2)
The revision addresses the comments of the reviewers and adds several new analyses and results. Among our findings was the discovery of significant batch effects that, respectively, differentiate gene expression of signature genes in the Discovery and Validation patient datasets. This is an important cautionary message that should be considered when analyzing the performance of any machine learning based method.
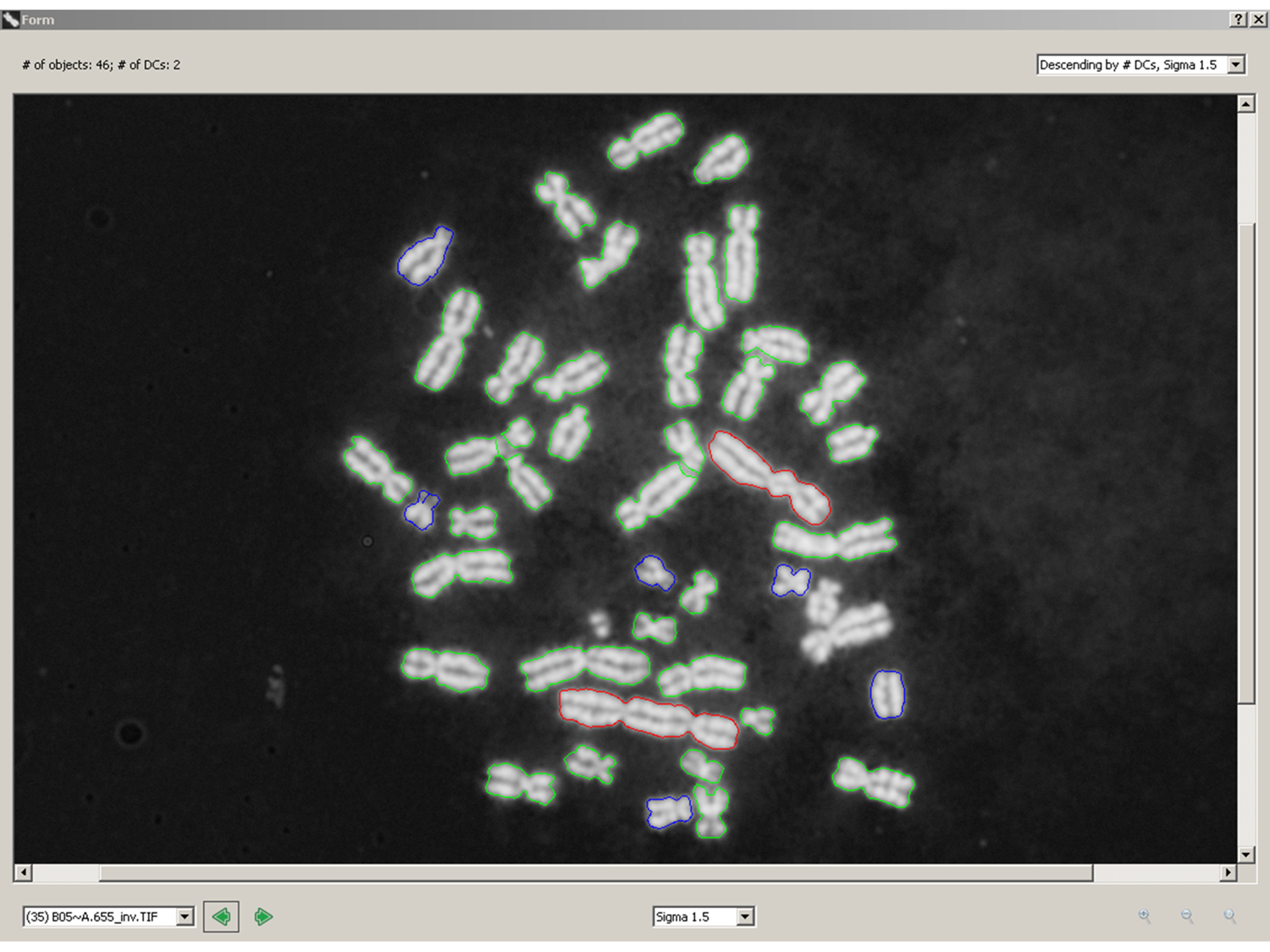
Counting pixel area and pixel intensities (stained antibodies, DNA or RNA) does not determine the identities of the cellular objects that are labeled. The challenge is that every microscope field exhibits different morphology, so traditional image segmentation algorithms aimed at identifying specific subcellular components may not be reliable. We need to be clever to ferret out generalizable image properties of specific cellular components, invariant to morphological variability, that will uniquely discriminate normal from abnormal subcellular distributions of the biomarker of interest. We have done this to identify dicentric chromosomes – see red objects in the attached figure (green are normal, monocentric chromosomes). It should be possible to do this for other subcellular objects. Contact CytoGnomix (mailto://info@cytognomix.com) to discuss further.
A postdoctoral position is available to work on a newly funded high-performance computing project:
“Automated Cytogenetic Dosimetry as a Public Health Emergency Medical Countermeasure.”
This 2 year project is supported by the SOSCIP-TalentEdge program. Candidates should be qualified in C++ development, preferably with experience in parallel computing. The position is at Western University in combination with the project partner Cytognomix.
Please contact either Drs. Knoll or Rogan if interested:
|
|
|
|
|
Once you see the discoveries that only MutationForecaster® can make, we are confident that you will sign up for a subscription to analyze your own data.
Contact us if you have questions.
Recently, we published 2 papers describing our unifying framework for non-coding mutation analysis (Mucaki et al. BMC Medical Genomic, 2016; http://bmcmedgenomics.biomedc
(Lu et al. 2016;http://nar.oxfordjournals.org/
I am not claiming that the variants we prioritize with our framework are definitively pathogenic, but do believe that strategies that are narrowly focused on the genetic code itself won’t advance the field or help patients much. Clinical molecular geneticists seriously consider sequencing beyond coding regions and trying to interpret the variants detected in the regions. The incremental costs to do this aren’t exorbitant, and the excuse of ignorance about the meaning of such variants is simply not valid any longer.
Many non-coding mutations have been proven ‘anecdotally’; studies have not been designed to determine the incidence of these types of mutations, in part due to the higher densities of variants in non-coding regions, identifying the clinically relevant ones is more daunting. This has been compounded by the lack of bioinformatic and genomic methods to generate a reliable and comprehensive and high throughput validation of variants outside of coding regions with adverse functional consequences . Suffice it to say, there are many individual reports in the published literature, but they are not generally being systemically uncovered because of the narrow focus on changes in coding regions that affect amino acid sequences.
The problem is not only where the variants reside, but an overly conservative philosophy that fails to consider other interpretations for the effects of variants, even within coding regions. It’s not just non-coding regions that contain missing pathogenic variants, but also coding variants where the change in the amino acid code may not be the source of the disease pathology. There are actually numerous examples of this phenomenon (and a number of good reviews eg. Cartegni et al (https://www.ncbi.nlm.nih.gov/
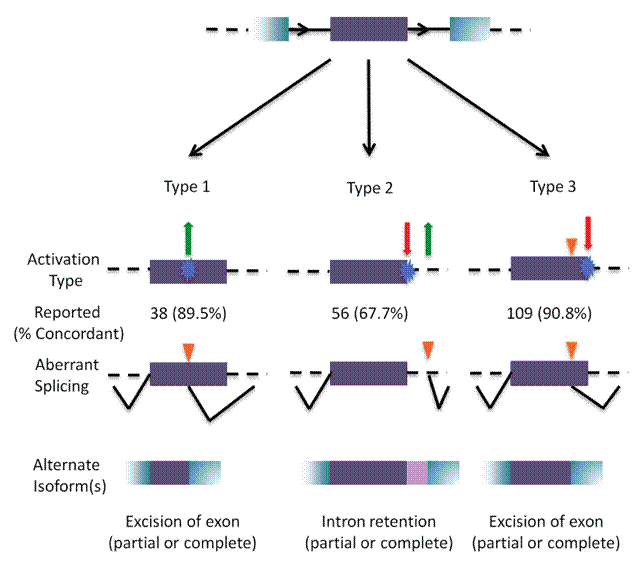
There is inevitably some bias against the reporting of intronic cryptic splicing mutations, because these sequences are not routinely determined in either research or clinical studies. Besides these classes, our studies also identify variants that alter transcription factor binding site strength and mRNA stability (in untranslated regions of mRNAs).
The exchange of mutation information about inherited breast cancer among various testing companies (except Myriad) has increased confidence in mutation interpretation. Those with rare mutations that are not shared among multiple patients do not benefit from this exchange. But these are generally based almost entirely on variants that cause amino acid substitutions or nonsense codons. I contend that such exercises, while very useful, are simply not scalable to the true volumes of all variants found in genes, and they ignore other mechanisms of pathogenicity such as those described above.
To reiterate, my argument is that current clinical molecular diagnostic practices will continue to leave many patients without known pathogenic mutations. Until this point of view changes and we seriously focus on functional and bioinformatic methods to analyze and prioritize VUSs thoughout genes, there will be a lot of frustration about the lack of results among the companies, academics and the patients they are purporting to help. We should also question whether the cost of testing can be justified, with the knowledge that a significant amount of genetic real estate is not being sequenced nor interpreted.
Peter K. Rogan
Cytognomix receives contract from the Build in Canada Innovation Program from the Government of Canada to test our novel ADCI software to estimate effects of exposure to ionizing radation. The project will be a collaboration with Health Canada and Canadian Nuclear Laboratories. ADCI determines the biological dose received without manual review and is suitable for evaluation of exposures in a mass casualty event.
US Patent No. 8,605,981 on CytoGnomix’s centromere finding algorithm, which is a key component of the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) software, was awarded in 2013. On November 8th 2016, German patent application No. 11 2011 103 687.6 on the same invention was granted as Patent No. 11 2011103687. We note that both of the major manufacturers of automated cytogenetic image capture systems are German and we look forward to working with them.


