The trademark on CytoGnomix MutationForecaster® system has been renewed for another 10 years.

The trademark on CytoGnomix MutationForecaster® system has been renewed for another 10 years.
Mucaki, EJ, Shirley BC, and PK Rogan.Improved radiation expression profiling by sequential application of sensitive and specific gene signatures. BioRxiv https://doi.org/10.1101/2021.08.18.456812
#artificialintelligence #radiationsafety #geneexpression #genesignature
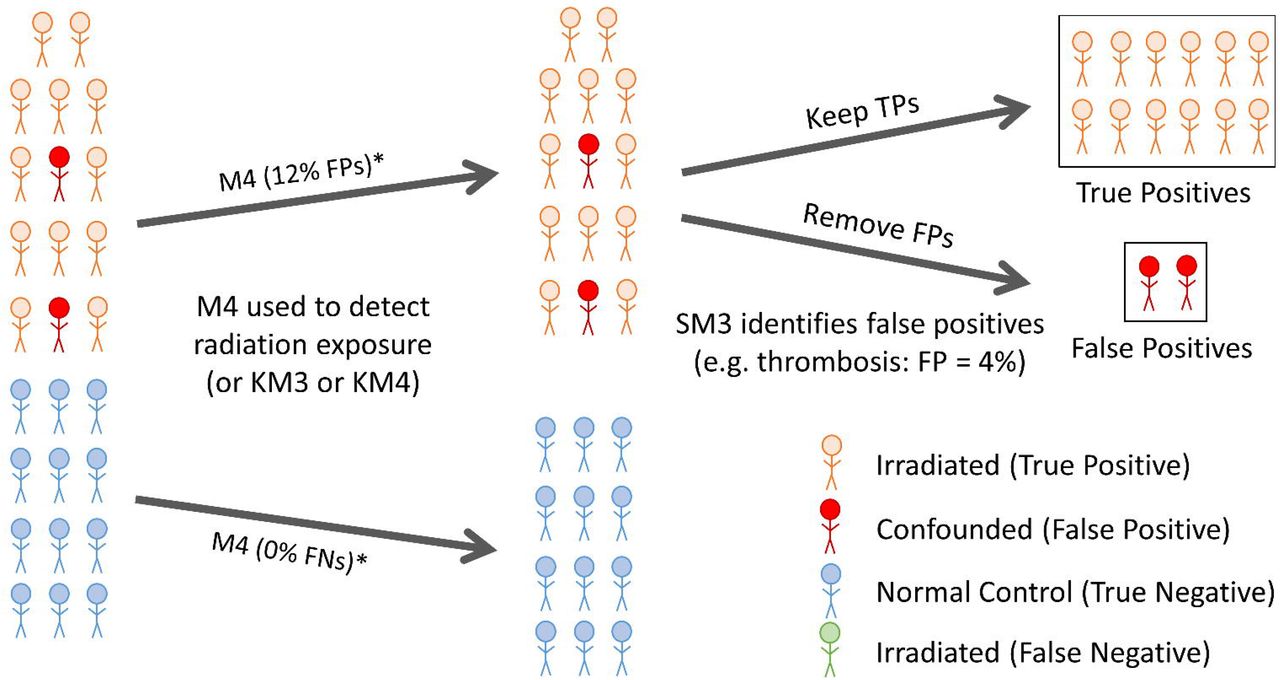
CytoGnomix will be presenting: “Radiation biodosimetry exposure assessment from gene expression signatures can be confounded by other underlying disease pathologies” at the CSA / NRC-IRAP Colloquium Healthcare Without Boundaries on June 2, 2021.

#medicine #healthcare #health #research #radiationprotection
On May 10, 2021, CytoGnomix is presenting a poster at ConRad 2021 (www.radiation-medicine.de) titled:
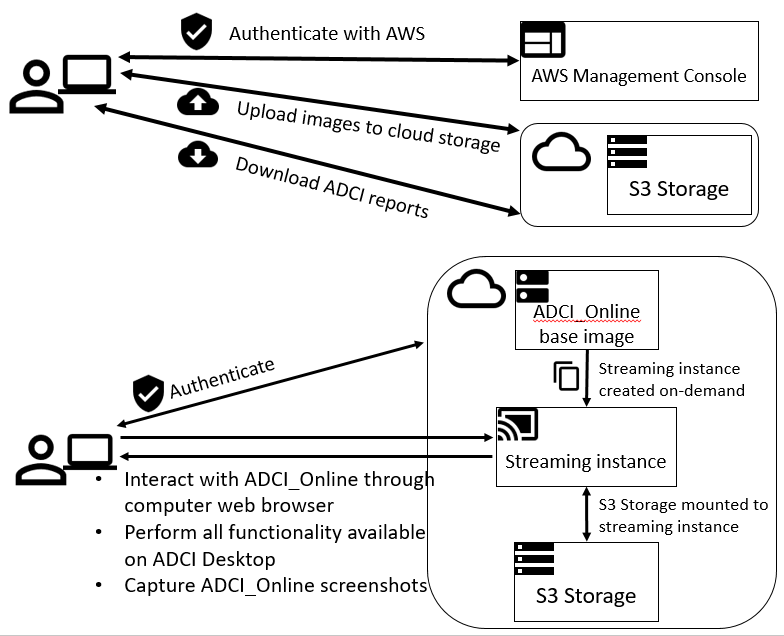
Demonstration of the Automated Dicentric Chromosome Identifier and Dose Estimator [ADCI] System in a Cloud-based, Online Environment.
From the abstract:
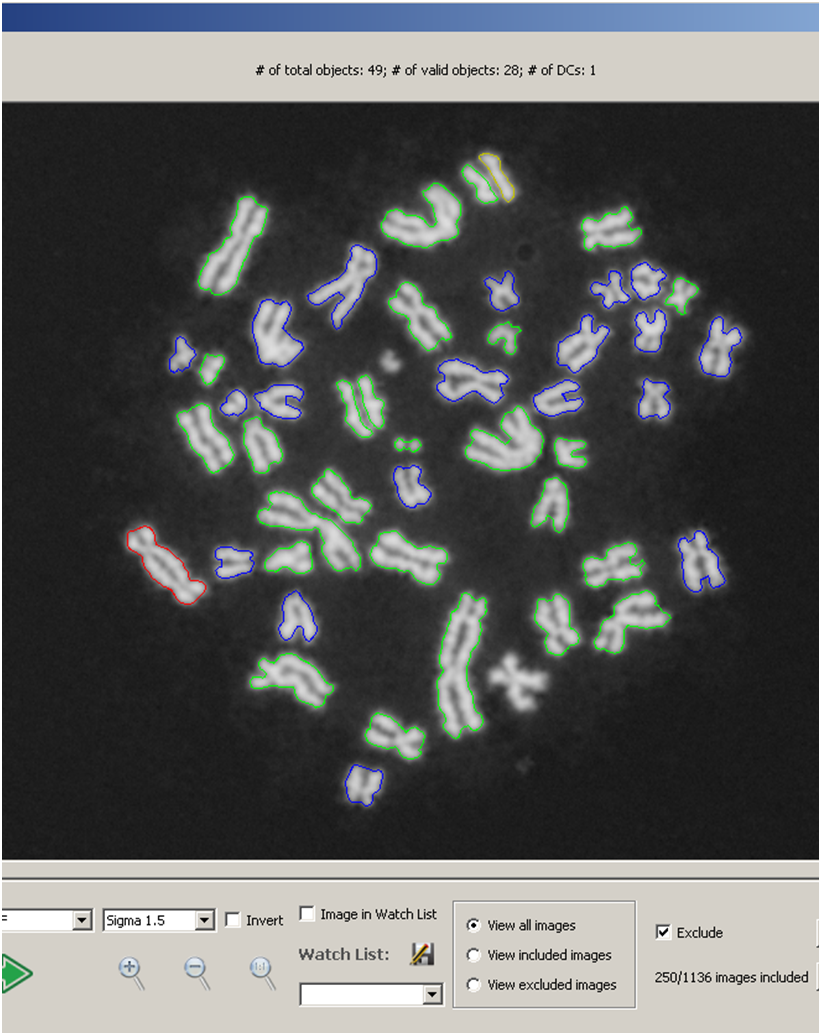
Interpretation of cytogenetic metaphase images and quantification of exposures remain labour intensive in radiation biodosimetry, despite computer-assisted dicentric chromosome (DC) recognition and strategies to share workloads among different biodosimetry laboratories. ADCI processes the captured images to identify DCs, selects images, and quantifies radiation exposure. This paper describes ADCI_Online, a secure web-streaming platform on Amazon Web Services that can be accessed worldwide from distributed local nodes.
ADCI_Online offers a subscription-based service useful for radiation research, biodosimetry proficiency testing, inter-laboratory comparisons, and training. In a research context, the system could provide highly uniform, reproducible assessment in large studies of many individuals, for example, exposed to therapeutic radiation. ADCI_Online compute environments originate from a single snapshot which can be cloned any number of times; thus, the system can be rapidly scaled when required. With robust network connectivity in a medical emergency of multiple potentially radiation exposed individuals, throughput and capacity for multiple samples requiring simultaneous processing and dose evaluation can be expanded to seamlessly mitigate any backlog in sample interpretation.
Rogan PK, Mucaki EJ, Shirley BC, Li Y, Wilkins RC, Norton F, Sevriukova O, Pham N-D, Waller E, Knoll JHM. Automated Cytogenetic Biodosimetry at Population-Scale. Radiation. 2021; 1(2):79-94. doi: 10.3390/radiation1020008 (2021)
CytoGnomix’s first #patent for automated interpretation of #radiation #biodosimetry exposures issued today.
(http://patft.uspto.gov/netacgi/nph-Parser?Sect1=PTO1&Sect2=HITOFF&d=PALL&p=1&u=%2Fnetahtml%2FPTO%2Fsrchnum.htm&r=1&f=G&l=50&s1=10929641.PN.&OS=PN/10929641&RS=PN/10929641)
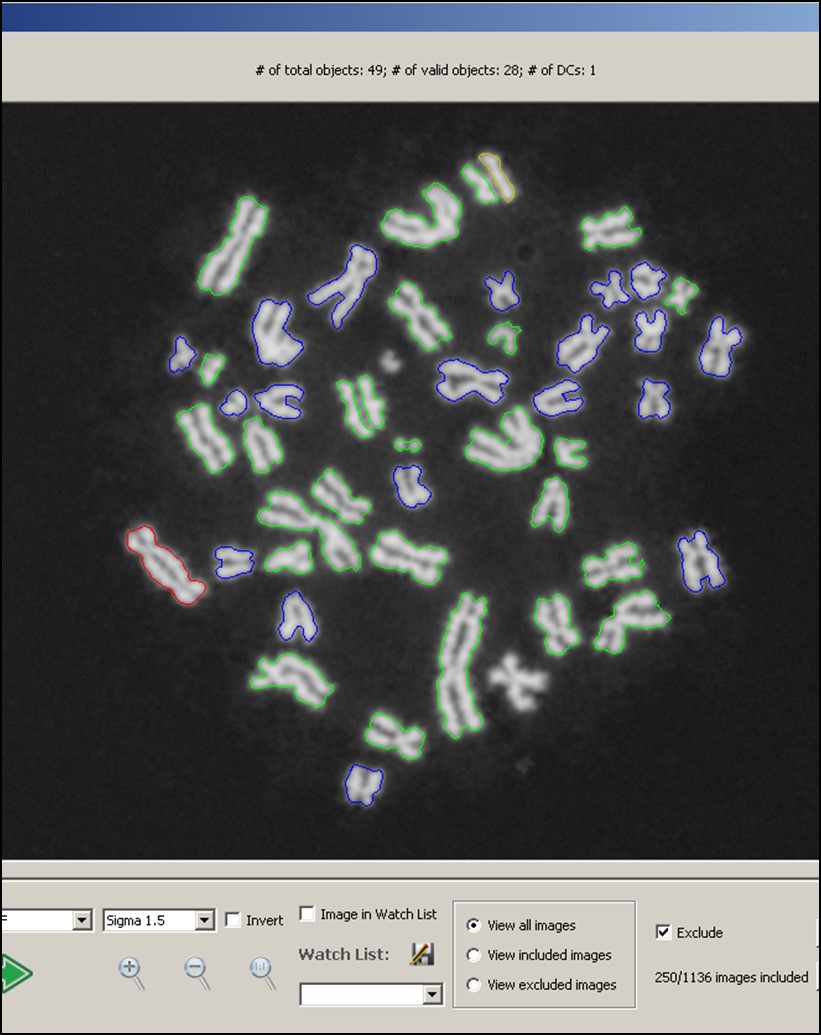
“Demonstration of the Automated Dicentric Chromosome Identifier and Dose Estimator System (ADCI™) in a Cloud-based Online Environment”
at the International Atomic Energy Agency Coordinated Research Project (CRP) E35010: Applications of Biological Dosimetry Methods in Radiation Oncology, Nuclear Medicine, Diagnostic and Interventional Radiology (MEDBIODOSE)
Greetings to you for a safe and healthy New Year.
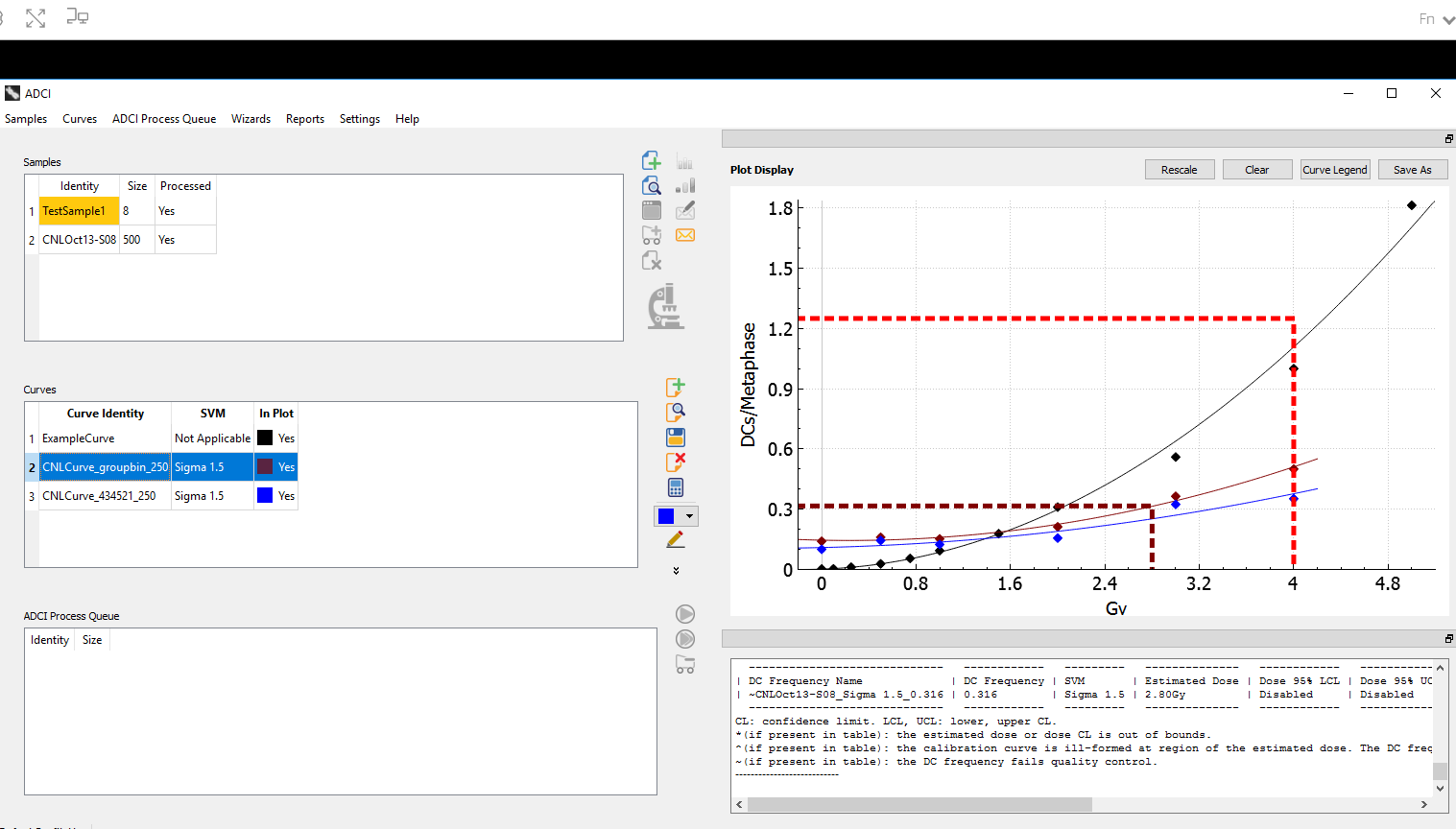
The Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) has become the biodosimetry industry’s leading software system for accurate and rapid quantification of absorbed ionizing radiation. This year we upgraded our Windows-based system to also determine partial body exposures, both fraction of cells exposed and whole body equivalent dose levels (Shirley et al. 2020).
In the coming year, CytoGnomix will introduce ADCI in the Cloud. This version of our software will make ADCI available as a highly secure web-application. All of the same functionality found in the Windows software will be available in ADCI_Online , except users will upload metaphase images to our AWS application. We have already validated the Demonstration Version of ADCI in this virtual environment. It is no longer necessary to download and install this software on your own computer in order to test drive it.
Contact us to access a Demo of ADCI_Online.
MedComm (Wiley) 1(3): 311-327, 2020. (https://doi.org/10.1002/mco2.46)
CytoGnomix’s Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) system will be awarded a US Patent for all claims covering “Smart Microscope System for Radiation Biodosimetry.” The patent application is available at:
https://patents.google.com/patent/US20200050831A1
The abstract reads:
An automated microscope system is described that detects dicentric chromosomes (DCs) in metaphase cells arising from exposure to ionizing radiation. The radiation dose depends on the accuracy of DC detection. Accuracy is increased using image segmentation methods are used to rank high quality cytogenetic images and eliminate suboptimal metaphase cell data in a sample based on novel quality measures. When a sufficient number of high quality images are detected, the microscope system is directed to terminate metaphase image collection for a sample. The microscope system integrates image selection procedures that control an automated digitally controlled microscope with the analysis of acquired metaphase cell images to accurately determine radiation dose. Early termination of image acquisition reduces sample processing time without compromising accuracy. This approach constitutes a reliable and scalable solution that will be essential for analysis of large numbers of potentially exposed individuals.
In response to the Call for Proposals under its Innovation for Defence Excellence and Security (IDEaS) program to address COVID-19 challenges, the Department of National Defence has recommended CytoGnomix’s project:
Locating emerging COVID19 hotspots in Ontario after community transmission by time-correlated, geostatistical analysis
for funding following evaluation against mandatory, point rated criteria, and strategic considerations.
The project will “provide financial support through a non-repayable Contribution Agreement up to $200,000 for the development of your proposed solution for a maximum performance period of six months.”
We have added the capability to determine whether samples exposed to ionizing radiation are wholly or partially irradiated. If partially, the approach determines the fraction of metaphase cells exposed and the whole body-equivalent dose completely automatically. CytoGnomix’s Automated Dicentric Chromosome Identifier and Dose Estimation software has been upgraded to generate these results as part of the the dose estimation report. The article has been accepted for publication by the International Journal of Radiation Biology V. 96 (https://doi.org/10.1080/09553002.2020.1820611). It is also currently available on BioRxiv:
Dr. Peter Rogan is presenting our laboratory’s recent F1000Research publication (9: 943) on a proposed molecular mechanism for pathogenesis of severe RNA-viral pulmonary infections at the 2020 RiboClub Forum:

#sarscov2 #covid19research #genomics #molecularmechanism #RNA #virology #covidー19
We are giving a platform presentation at the upcoming American Society of Human Genetics virtual meeting #ASHG2020
Session: Personalized Medicine Approaches in Healthcare.
Paper: Pathway-extended gene expression signatures integrate novel biomarkers that improve predictions of responses to kinase inhibitors
P. K. Rogan(1,2), A. J. Bagchee-Clark(1), E. J. Mucaki(1). 1. Department of Biochemistry, University of Western Ontario and 2. CytoGnomix Inc., London ON Canada

Abstract: Individualized chemotherapy selection in cancer potentially maximizes drug efficacy while minimizing drug toxicity. Despite the knowledge of many pharmacogenetic biomarkers, inter-individual variability in response to chemotherapeutic response has limited the success of the approach. We derive multi-gene expression signatures that predict individual patient responses to tyrosine kinase inhibitors (TKIs): erlotinib, gefitinib, sorafenib, sunitinib, lapatinib and imatinib. Gene models for TKIs implicated from the published literature tend to predict either sensitivity or resistance to TKIs well (but not both). This
issue was addressed with a systems biology-based strategy that expanded with candidate gene products related to these genes in these models through biochemical pathways and interactions. Using patient transcriptome data, these Pathway-Extended (PE) models predicted responses for individual patients that matched observed outcomes at accuracies of 65% (imatinib), 71% (lapatinib and gefitinib), 78% (sunitinib), 83% (erlotinib) and 89% (sorafenib). After training and evaluating many extended signatures, those with the strongest predictive performance were composed primarily of pathway-related genes that according to post-hoc analysis were clearly implicated in cancer phenotypes. Machine learning-based PE expression signatures display strong efficacy in predicting both sensitivity and specificity in patients through incorporation
of novel cancer biomarkers.
#genomics #medicine #chemotherapy #cancertreatment #kinaseinhibitors
Peter Rogan was interviewed by Gill Eapin for his daily podcast, Scientific Sense, focused on Science & Economics about our research projects about COVID19. Listen at the link below.

#medicine #sarscov2 #covidー19 #medicalsciences #epidemiology #health #geostatistics #hotspots #publichealth #genomics #molecularmechanism
We have described and provide evidence for an explanation for how rapid onset, severe RNA viral infections, such as SARS-CoV-2 or Influenza, may develop:
Rogan PK, Mucaki EJ and Shirley BC. A proposed molecular mechanism for pathogenesis of severe RNA-viral pulmonary infections. F1000Research 2020, 9:943 (https://doi.org/10.12688/f1000research.25390.1)