PgmNr 182: Splicing mutation risk analysis in hereditary breast and ovarian cancer exomes. (Platform)
Thurs, Oct 19. 11:00am -12:30pm. Session 40. Defining High Risk in Cancer. Room 230C – Level 2/Orlando Convention Center
E.J. Mucaki 1; B.C. Shirley 2; S.N. Dorman 1; P.K. Rogan 1,2 1) Biochemistry, University of Western Ontario, London, Ontario, Canada; 2) CytoGnomix Inc, London, Ontario, Canada
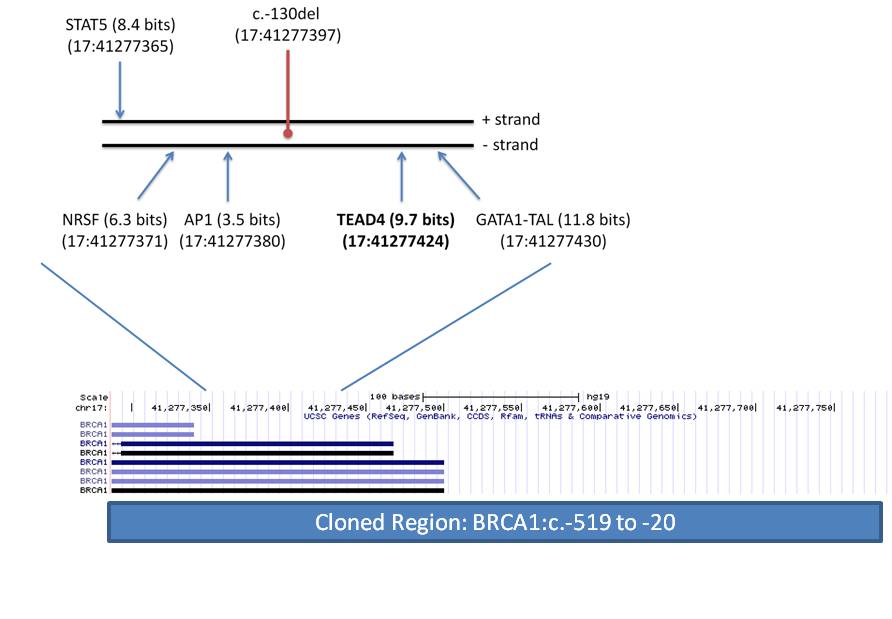
Genetic testing of patients with inherited cancer frequently reveals variants of unknown significance (VUS). We have presented an Information Theory (IT) framework to predict and prioritize coding and non-coding VUS in hereditary breast and ovarian cancer (BRCA) patients, including effects on mRNA splicing1,2. We investigated the exome wide distribution of predicted mRNA splicing mutations in a large BRCA cohort. Predicted splicing mutations in IT-based splicing analysis of all variant data from AmbryShare BRCA exome (n=11,416; with 1.2 million VUS) and the control genome Aggregation Databases (gnomAD; n=138,632) were identified using the Shannon splicing mutation software pipeline3. IT-flagged variant frequencies (decreasing Ri values [in bits] of either leaky or inactivated natural splice sites [∆Ri >4 bits and Ri ≤ 1.6] or strengthened cryptic splices sites with an Ri exceeding that of adjacent natural sites) were compared for each gene using odds ratios (OR). ORA is defined as the ratio of frequencies of the same flagged variants in a gene in AmbryShare relative to gnomAD. ORP is based on the ratio of frequencies of all flagged variants in a gene in AmbryShare relative to all flagged variants in that gene in gnomAD. A greater number of IT-flagged variants were present in AmbryShare than in gnomAD among 2012 genes with severe splicing mutations. Increasing the ∆Ri threshold disproportionally decreases the number of flagged variants in gnomAD due to fewer severe splicing mutations. Variants that abolish natural splice sites flagged known inherited breast cancer genes with respectively increased ORA and ORP inATM (493, 407), BARD1 (407, 407), BRCA1 (19, 14), BRCA2 (54, 54),CDH1 (549, 549), MLH1 (303, 303), MUTYH (95, 11), and PALB2 (233, 116). Other flagged breast cancer-related genes with high OR includeAAMP, C1QTNF6, CDK3, FOLR1, PRLR, RAD50, RING1, S100A2, SRGN,TMSB10, TYRO3, and VIM. Notable highly mutated genes from other cancers include GKN1 (gastric), C1orf61 (hepatocellular), CREM(prostate), PNKP (multiple), PPP1CA (gastric) and ZFAND2B (myeloid). Flagged genes not known to be linked to cancer include ATP1A4, MFF,PACSIN1, PTS, and USH1C. Severe splicing mutations occur more frequently in inherited and somatic breast cancer genes as well as in other genes in BRCA populations.
1Mucaki et al. BMC Med. Genom. 9:19, 2016; 2Caminsky et al. Hum. Mut. 37:640, 2016; 3Shirley et al. Genom. Prot. Bioinf. 11:75, 2013. Keywords: Cancer; Bioinformatics; Genomics; Population genetics; Statistical genetics
PgmNr 1268/T: Accurate radiation biodosimetry through automation of metaphase cell image selection and chromosome segmentation. (Poster)
Thurs, Oct 19. 2:00pm – 4:00pm. Bioinformatics and Computational Approaches. Exhibit Hall, Level 1, Orlando Convention Center
Y. Li 1; J. Liu 2; B. Shirley 1; R. Wilkins 3; F. Flegal 4; J.H.M. Knoll 1,2; P.K. Rogan 1,2 1) CytoGnomix Inc, London, Ontario, Canada; 2) University of Western Ontario, London, Ontario Canada; 3) Health Canada, Ottawa, Ontario, Canada; 4) Canadian Nuclear Laboratories, Chalk River, Ontario, Canada
The dicentric chromosome (DC) assay is a standardized method that is recommended for determination of biologic radiation exposure1,2. Software to fully automate this assay has been developed in our laboratory3. This method relies on high quality microscope-derived images of metaphase cells to reduce the rate of false positive (FP) DCs. We present image processing methods to eliminate suboptimal metaphase cell images based on novel quality measures and to reclassify FPs by analyzing their morphological features. A set of chromosome segmentation thresholds selectively filtered out FPs, arising primarily from extended prometaphase chromosomes, sister chromatid separation and chromosome fragmentation. This reduced the number of FPs by 55% and was highly specific to the abnormal structures (≥97.7%). Image segmentation filters selectively remove images with consistently unparsable or incorrectly segmented chromosome morphologies, while image ranking sorts images according to their qualities and enables selection of optimal images in samples. Overall, these methods can eliminate at least half of the FPs detected by manual image review. By processing data to derive calibration curves and to assess samples of unknown exposures with the same image selection models, average dose estimation errors were reduced from 0.6 Gy to 0.3 Gy, without requiring manual review of DCs. During this presentation, we will use our software to demonstrate that metaphase image filtering and object selection constitute a reliable and scalable approach for biodosimetry, resulting in more accurate radiation dose estimates.
1. International Atomic Energy Agency. (2001) Cytogenetic Analysis for Radiation Dose Assessment, a Manual: Technical Reports Series. No. 405, International Atomic Energy Agency, Vienna.
2. International Atomic Energy Agency. (2011) Cytogenetic Dosimetry: Applications in Preparedness for and Response to Radiation Emergencies, International Atomic Energy Agency, Vienna.
3. Rogan, P. K., Li, Y., Wilkins, R. C., Flegal, F. N., and Knoll, J. H. M. (2016) Radiation Dose Estimation by Automated Cytogenetic Biodosimetry, Radiation Protection Dosimetry 172, 207-217.
Keywords: Bioinformatics; Centromere structure/function; Chromosomal abnormalities; Diagnostics; Public health
PgmNr 1288/W: Predicting exposure to ionizing radiation by biochemically-inspired genomic machine learning. (Poster)
Wed, Oct 18. 3:00pm – 4:00pm. Bioinformatics and Computational Approaches. Exhibit Hall, Level 1, Orlando Convention Center.
J.Z.L. Zhao; E.J. Mucaki; P.K. Rogan. Dept Biochemistry, University of Western Ontario, and CytoGnomix Inc., London, Ontario, Canada
Analyzing gene expression in peripheral blood mononuclear cells reveals profiles that predict radiation exposure in humans and mice by logistic regression (PLoS Med. 4:e106; PLoS ONE. 3:e1912). Using biochemically-inspired methods (Mol. Onc. 10:85-100), we derive gene signatures to predict the level of radiation exposure with improved accuracies. DNA repair genes responsive or differentially expressed upon radiation exposure and orthologs highly expressed in species resilient to radiation exposure (n=998) were analyzed by two-sampled t-tests comparing expression in individuals unexposed and exposed to radiation (150-200 cGy: humans or 50-1000 cGy: mice). Significance thresholds for including a gene in developing a signature were adjusted based on radiation dose, from p < 0.01 (50 cGy) to < 1E-14 (1000 cGy), equivalent to ~10% of genes. Support Vector Machine (SVM) signatures were derived by backward feature selection (BFS) or minimum-redundancy-maximum-relevance (mRMR) and validated using leave-one-out cross validation (LOOCV) and external datasets. GEO datasets GSE6874 and GSE10640 were used for training and testing. Signatures derived by BFS from the human patients of GSE6874 (n=78) included α) GADD45A, GTF3A, TNFRSF4, XPC and β) ATR, GADD45A, GTF3A, IL2RB, MYC, NEIL2, RBM15, SERPINB1, XPC, which both distinguished irradiated from unirradiated individuals with 98% sensitivity and 100% specificity in LOOCV. Validating these signatures on the human patients of GSE10640 (n=71) confirms that α and β are both 92% sensitive and, respectively, 94% and 96% specific. mRMR found the 10 “best” genes from the murine samples of GSE10640 (n=104) to create a signature at each radiation dose; several genes were common among signatures. Signature δ (50 cGy) included PHLDA3, BAX, NBN, CCT3, CDKN1A, CCNG1, POLK, ERCC5, GCDH, and RAMP1. Signature ε (200 cGy) included PHLDA3, LIMD1, CCT3, BAX, MS4A1, GLIPR2, BLNK, BCAR3, CDKN1A, andTFAM. Signature ζ (1000 cGy) included CCT3, SUCLG2, EI24, CNBP, PHLDA3, TPST1, HEXB, FEN1, CDKN1A, and BLNK. When validated on the murine samples of GSE6874 (n=14), each signature correctly predicted the exposure status of all mice. Our approach produces signatures with higher accuracies in cross- and external validation datasets than prior logistic regression models, with significantly improved sensitivities in detecting radiation exposure in humans. This will be useful in identifying nearly all radiation-exposed individuals in a mass casualty.
Keywords: Bioinformatics; Diagnostics; Transcriptome; Computational tools; Hematopoietic system