Population scale biodosimetry with the Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) software system. (Platform) Rogan PK, Ali, S, Li Y, Shirley B, Wilkins R, Flegal F, Cooke R, Peerlaproulx T, Waller E, Knoll JHM. EPR Biodose 2018, June 11-15, Munich Germany
Optimization of image selection in Automated Dicentric Chromosome Analysis. Li Y, Shirley B, Wilkins R, Flegal, F, Knoll JHM, Rogan PK. EPR Biodose 2018, June 11-15, Munich Germany.
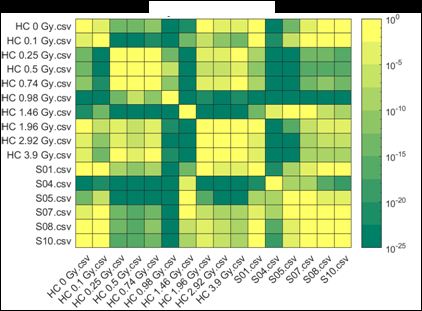
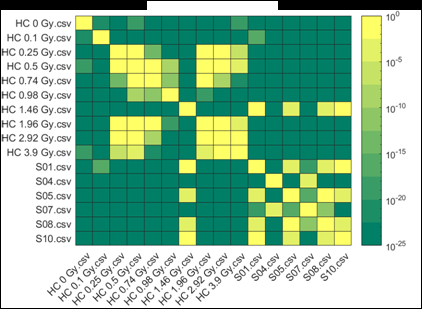
Predicting exposure to ionizing radiation by biochemically-inspired genomic machine learning. Rogan PK, Zhao JZL, and Mucaki EJ. EPR Biodose 2018, June 11-15, Munich Germany.

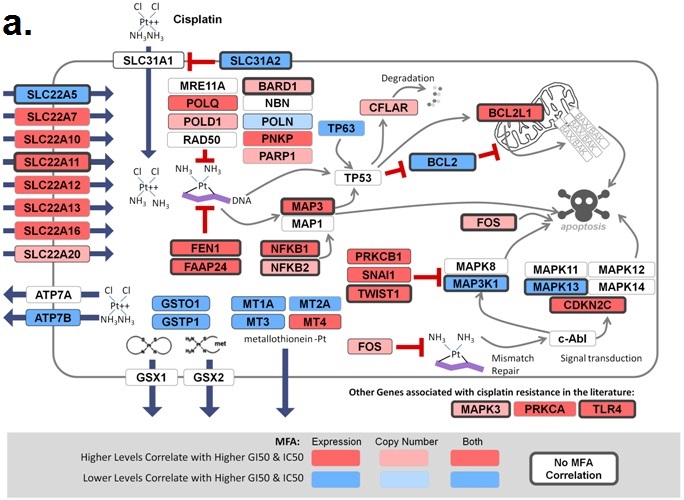
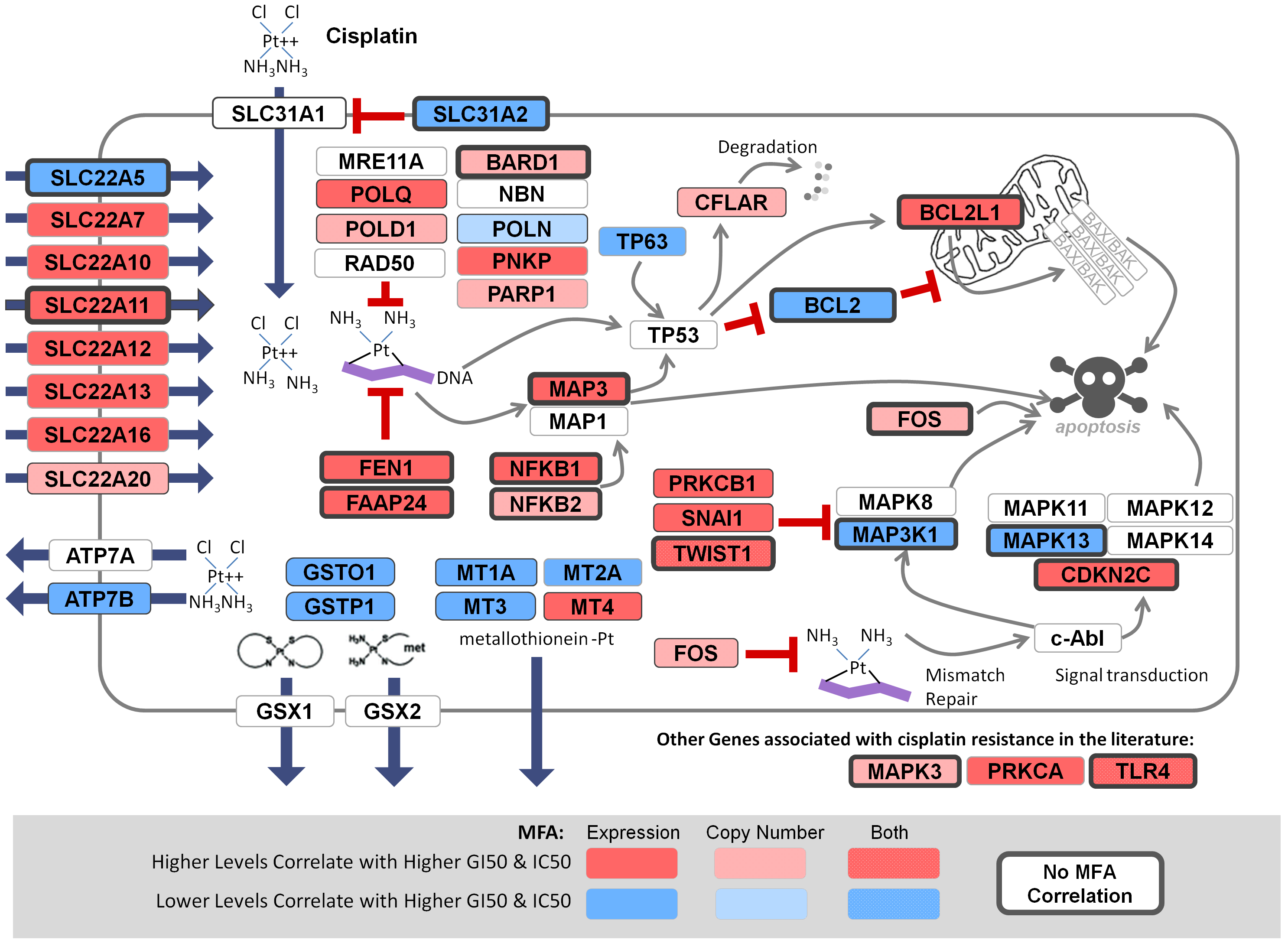
Comprehensive prediction of responses to chemotherapies by biochemically-inspired machine learning. (Best Poster session) Rogan PK, Zhao JZL, Mucaki EJ. European Society of Human Genetics 2018, June 16-19, Milan Italy.