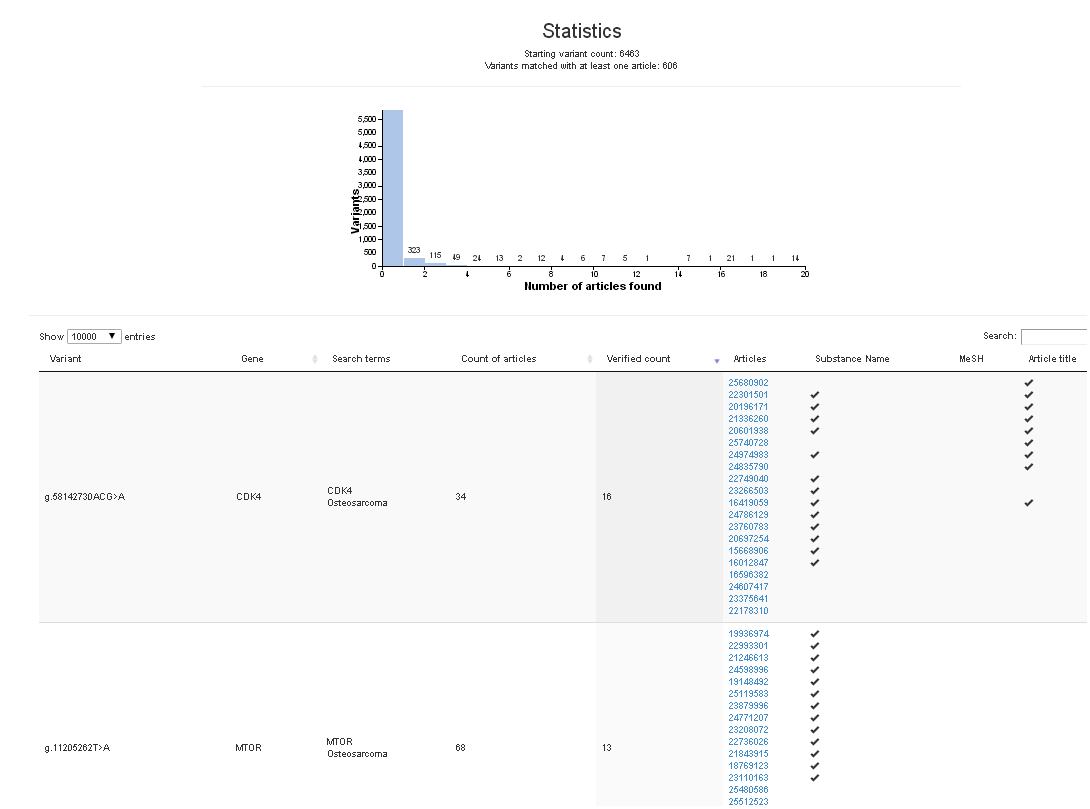
Paper: Caminskyetal_2016 uncorrectedproof


Our new paper on interpretation of gene variants in inherited breast and ovarian cancer has been accepted for publication in the journal, Human Mutation as a Research Article.
“Prioritizing variants in complete Hereditary Breast and Ovarian Cancer (HBOC) genes in patients lacking known BRCA mutations,” by Natasha G. Caminsky1, Eliseos J. Mucaki1, Ami M. Perri1, Ruipeng Lu2, Joan HM. Knoll3,4 and Peter K. Rogan1,2,4,5.
1Department of Biochemistry, Schulich School of Medicine and Dentistry, Western University, London, Canada, N6A 2C1, 2Department of Computer Science, Faculty of Science, Western University, London, Canada, N6A 2C1, 3Department of Pathology and Laboratory Medicine, Schulich School of Medicine and Dentistry, Western University, London, Canada, N6A 2C1, 4Cytognomix Inc. London, Canada, 5Department of Oncology, Schulich School of Medicine and Dentistry, Western University, London, Canada, N6A 2C1
A preprint of this article is published at http://biorxiv.org/content/early/2016/02/09/039206
We have added new capabilities to Variant Effect Predictor. Exome sequencing reveals many variants that have little or no effect on phenotype. You can remove these variants in MutationForecaster with our new stringency filters. Different default levels of filtering are offered. These can also be customized based on allele frequencies, predicted SIFT, Polyphen, variant type (eg. synonymous change), or protein coding domain containing the variant.
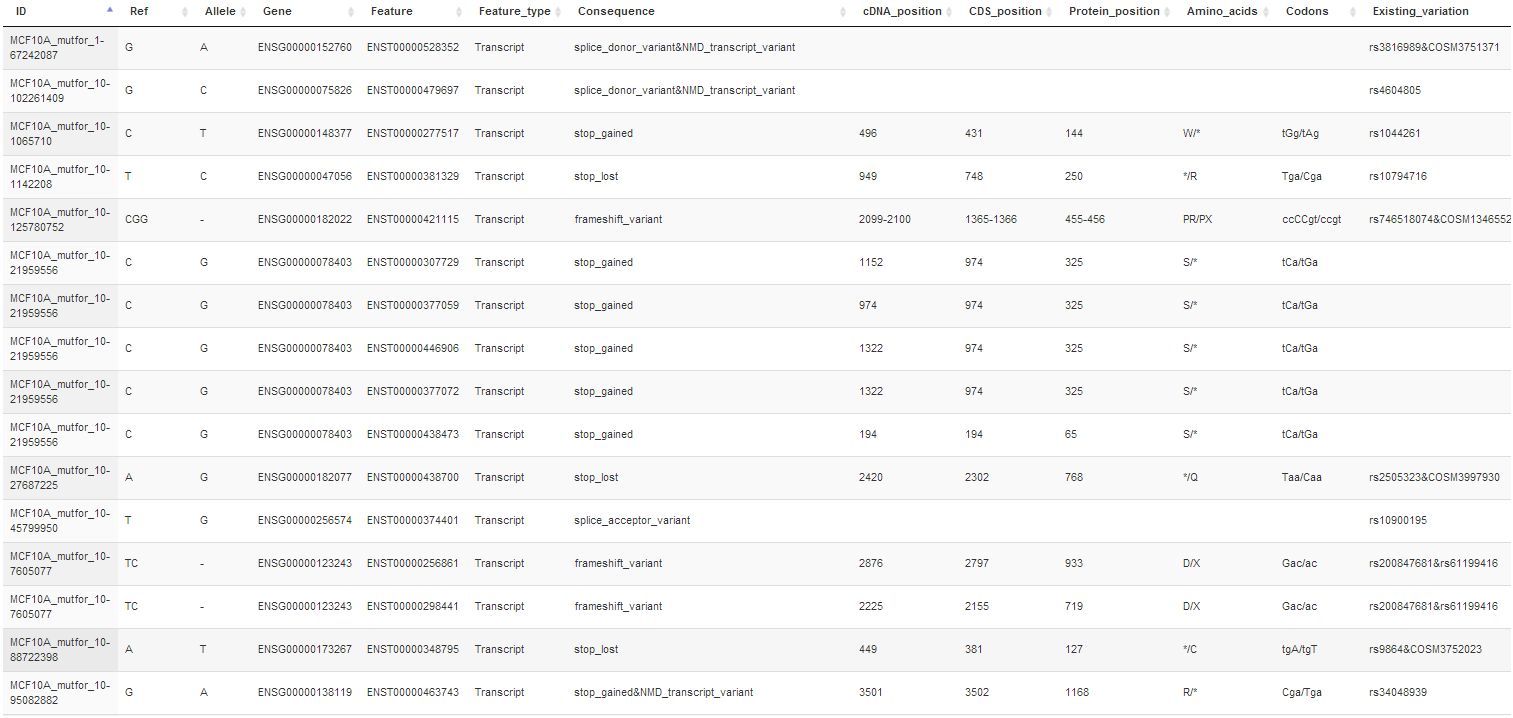
Yanxin Li1, Joan H. Knoll2,3, Ruth Wilkins4, Farrah N. Flegal5, and Peter K. Rogan1,3* Automated Discrimination of Dicentric and Monocentric Chromosomes by Machine Learning-based Image Processing. Departments of 1Biochemistry, and 2Pathology and Laboratory Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, 3Cytognomix Inc., 4Health Canada, and 5Canadian Nuclear Laboratories.
in the journal Microscopy Research and Technique.
Abstract: Dose from radiation exposure can be estimated from dicentric chromosome (DC) frequencies in metaphase cells of peripheral blood lymphocytes. We automated DC detection by extracting features in Giemsa-stained metaphase chromosome images and classifying objects by machine learning (ML). DC detection involves i) intensity thresholded segmentation of metaphase objects, ii) chromosome separation by watershed transformation and elimination of inseparable chromosome clusters, fragments and staining debris using a morphological decision tree filter, iii) determination of chromosome width and centreline, iv) derivation of centromere candidates and v) distinction of DCs from monocentric chromosomes (MC) by ML. Centromere candidates are inferred from 14 image features input to a Support Vector Machine (SVM). 16 features derived from these candidates are then supplied to a Boosting classifier and a second SVM which determines whether a chromosome is either a DC or MC. The SVM was trained with 292 DCs and 3135 MCs, and then tested with cells exposed to either low (1 Gy) or high (2-4 Gy) radiation dose. Results were then compared with those of 3 experts. True positive rates (TPR) and positive predictive values (PPV) were determined for the tuning parameter, sigma. At larger sigma, PPV decreases and TPR increases. At high dose, for sigma= 1.3, TPR = 0.52 and PPV = 0.83, while at sigma= 1.6, the TPR = 0.65 and PPV = 0.72. At low dose and sigma = 1.3, TPR = 0.67 and PPV = 0.26. The algorithm differentiates DCs from MCs, overlapped chromosomes and other objects with acceptable accuracy over a wide range of radiation exposures.
A preprint of the paper is available at bioRxiv: http://biorxiv.org/content/early/2016/01/19/037309
We have made some major improvements to the Cytognomix User Variation Database recently. These are described in this recent video by Shannon Brown, a software developer at our company: CUVD Video
MutationForecaster now generates comprehensive genome interpretation on-the-fly. The results from all of our gene variant interpretation modules (Shannon Splicing Mutation Pipeline, ASSEDA, VEP, and Veridical) can now be automatically processed by CytoVA to find mutated genes in the genome related to a particular phenotypes based on published literature. Results are also be immediately processed to find dysfunctional biochemical pathways common to multiple mutated genes. All of the results are directly imported to your own CUVD repository, where all the results for each variant are grouped together.
The process is completely unattended. Start the Workflow for an variant set from an exome or genome sequence; several hours later all of the analyses are finished for you to review in your own CUVD database.
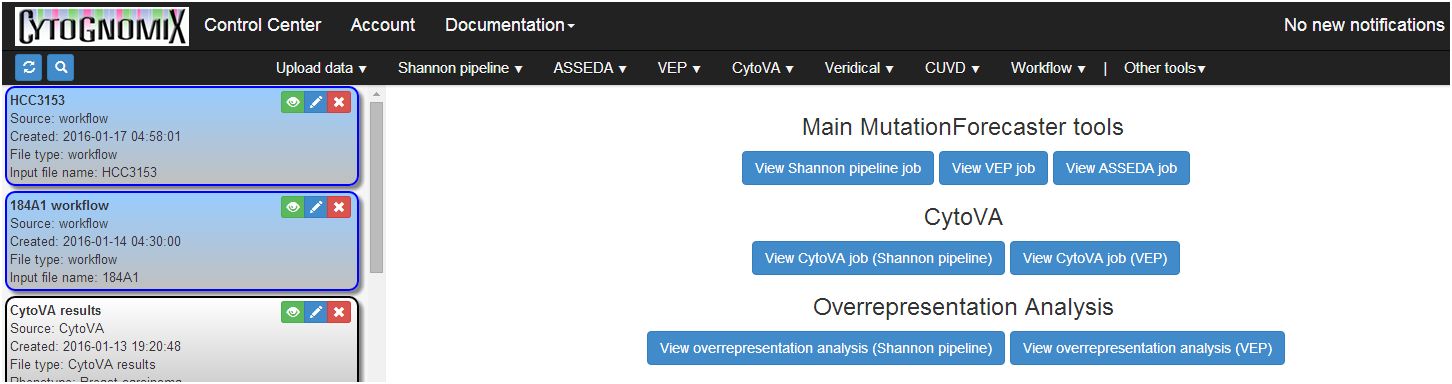
Every gene variant imported into CUVD from our other genome interpretation modules can be searched in several external databases seamlessly. Currently, all LOVD locus specific databases, dbSNP, ClinVar, and the Exome Variant Server are searched together and variants found in any of these resources are added to CUVD and hyperlinked when the search is completed. Until today, only one variant at a time could be searched.
As of today, CUVD is now able to simultaneously search and retrieve these data from batches of multiple variants with a single request (see below). Select all or just a group of variants in your database. MutationForecaster® estimates how long the search will take and notifies you when the task is complete. For example, searching 20 variants takes just over 1 minute. Replace outdated results when the databases are updated simply by repeating the search. Sign up for your free trial of MutationForecaster and try this exciting feature yourself!

The final version of our paper:
Dorman S, Baranova K, Knoll J, Urquhart, B, Marciani G, Carcangiu M-L, and Rogan PK. Genomic signatures for Paclitaxel and Gemcitabine resistance in breast cancer derived by machine learning. Mol. Oncology 10: 85-100, 2015. doi: 10.1016/j.molonc.2015.07.006
is available in print and here: Dorman etal Mol. Onc.10:85-100, 2016
We are excited to be able to offer our customers and registrants this opportunity to experience our integrated suite of genome interpretation products. For the first time, Cytognomix is offering a free trial of our MutationForecaster® genome interpretation suite to all registrants of the product. No subscription is required to analyze data with any of our software tools. Trial users are provided with the same datasets that we have analyzed in our peer-reviewed publications. Start your trial whenever you’re ready.
The trial showcases many capabilities available to subscribers:
2. Customize results with any of these products:
3. Streamline analysis of a dataset with all of these products in a single run using Workflows
Once you see the discoveries that only MutationForecaster® can make, we are confident that you will sign up for a subscription to analyze your own data.
Contact us if you have questions about the trial.
Happy holidays!
“Predicting response to adjuvant chemotherapy with genomic signatures derived by machine learning”
Department of Oncology, London Regional Cancer Program, University of Western Ontario.
The lecture room was packed and many oncologists attended!

The Splicing Mutation Calculator web software described in:
Caminsky NG, Mucaki EJ and Rogan PK. Interpretation of mRNA splicing mutations in genetic disease: review of the literature and guidelines for information-theoretical analysis [version 2; referees: 2 approved] F1000Research 2015, 3:282 (doi: 10.12688/f1000research.5654.2)
has been migrated to the MutationForecaster system (http://mutationforecaster.com). Subscribers to MutationForecaster have unlimited access to this product.
The one year free trial to this commercially-developed software has ended. The original website has been deprecated and no longer provides this functionality.
In next generation sequencing, exomes in particular, the challenge is to find relevant pathogenic gene variants among a sea of superfluous sequence changes. But the track record for filtering the most likely causative changes is dismal (20-25%). Most filtering methods remove common variants but do little else. Cytognomix has developed CytoVA, software that relates variants to patient peer-reviewed phenotypes in real time. We are adding this to our MutationForecaster system. Check it out!
Peter Rogan will present:
“Genomic analysis of metastasis and tumor chemotherapy response based on information theory and machine learning”
Department of Computer Science
University of Windsor
Date: Friday, November 13th, 2015
Time: 11:00 am
Location: Chrysler Hall – G100
Abstract: The integrated analyses of cancer phenotypes with complex genomic datasets has resulted in many new insights into diagnosis and prognosis. However, there is no single correct way to analyze these data, and the data themselves can vary significantly in content and interpretation between different studies of the same tumor type. We have used mutation, expression and copy number data to study breast cancer genes and genomes (hereditary and somatic). A major challenge in inherited breast cancer is the missing heritability; pathogenic mutations are not detected despite strong family historie. Our approach has been to prioritize functionally significant variants using information theory-based models of DNA and RNA binding protein binding sites. These same approaches – when applied to breast tumour exome sequences – have revealed numerous missed mRNA splicing mutations, and identified mutated pathways, validated by RNA sequencing, that are overrepresented in these tumour genomes. Application of biochemically-inspired machine learning to these integrated genomic data from cell lines produces gene signatures that robustly predict therapeutic response that we have validated with patient tumor data. Machine learning is a promising general approach that can be used for other drugs and tumor types with good recall.
Peter Rogan will be presenting:
Seeking the “Missing Heritability” in High-Risk Hereditary Breast and Ovarian Cancer (HBOC) Patients By Prioritizing Coding and Non-Coding Variants in 21 Genes. Natasha Caminsky G, Eliseos Mucaki J, Amelia Perri M, Ruipeng Lu, Matthew Halvorsen, Alain Laederach, Joan Knoll HM, Peter Rogan K
on Tuesday, November 10 from 12-2 PM in the poster session: Genomics, Proteomics, and Bioinformatics
in Montréal – Hôtel Bonaventure.
Scientific Program: link
Abstract:
Current BRCA1 and BRCA2 genetic testing for hereditary breast and ovarian cancer (HBOC) is often uninformative. The “missing heritability” may be due to variants in uninvestigated regions of these genes or variants in other genes. We have applied a unified framework based on information theory (IT) to predict and prioritize non-coding variants of uncertain significance. We captured complete gene sequences of 21 diseaserelevant genes in HBOC patients with uninformative hereditary predisposition testing (N=336) by hybridization enrichment using ab initio single copy probes that comprehensively span non-coding regions and flanking sequences of ATM, ATP8B1, BARD1, BRCA1, BRCA2, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51B, STK11, TP53, and XRCC2. We identified 38,538 unique variants. Eight were likely pathogenic BRCA1/2 mutations previously undetected by clinical testing. Eight proteintruncating mutations were identified in non-BRCA genes, the majority of which were in PALB2 (N=5), and 148 missense variants were flagged. Information weight matrices were derived for transcription factor (TFBS), splicing regulatory (SRBS), and RNA-binding (RBBS) protein binding sites from high-throughput sequencing data. IT analysis prioritized 12 variants affecting splicing (6 natural, 6 cryptic), 71 TFBS, 218 SRBS, and 29 RBBS. Co-segregation analysis found the relative risk of breast cancer for likely pathogenic BRCA variants torange from 1.55 to 75.78. According to clinically accepted guidelines, twenty-three were possibly pathogenic (13 confirmed by Sanger sequencing to date), 472 were of uncertain significance, and all remaining were likely not pathogenic. Complete gene analysis of BRCA1/2 and other genes is a successful strategy for identifying probable mutations in previously uninformative HBOC patients.
Ben Shirley, Chief software architect at Cytognomix, will be presenting:
Interpreting variants in complete gene and genome sequences with MutationForecaster®
at 11:50 AM at the Toronto NGS Symposium (Ben Sadowski Auditorium, 18th Floor, Mt Sinai Hospital, University Ave.).
Drs. Joan Knoll and Peter Rogan gave platform presentations about the underlying algorithms and application of the Automated Dicentric Chromosome Identifier and Radiation Dose Estimator:
“Radiation dose estimation by automated chromosome biodosimetry” and
at the EPRBiodose meeting at Dartmouth College, organized by the International Association of Biological and EPR Radiation Dosimetry .
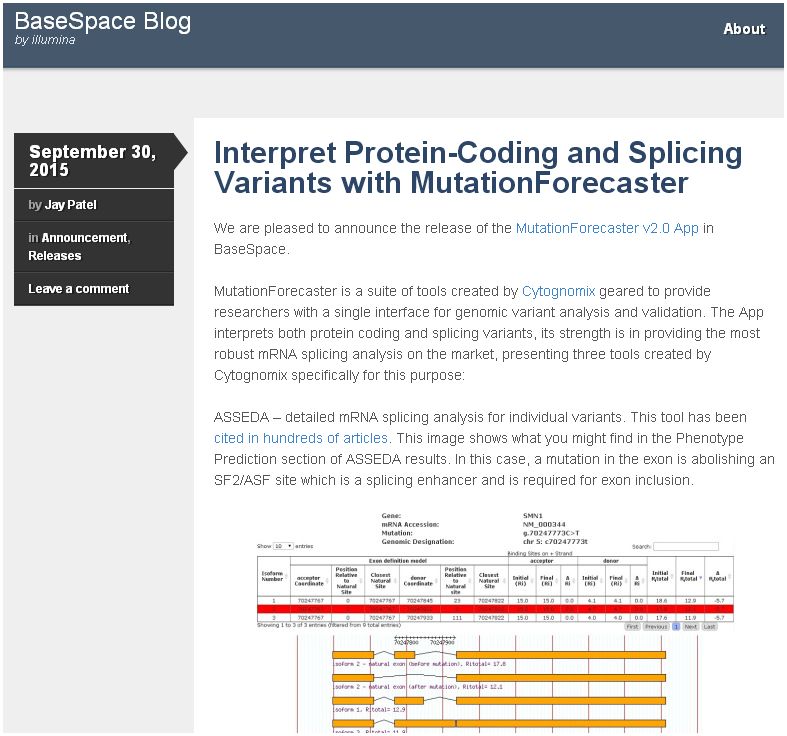
Please read our colorful blog entry describing the MutationForecaster app, now available on the Illumina® BaseSpace website (link). Short term subscriptions are available immediately from Illumina.
Peter Rogan and Katherina Baranova were interviewed by Jan Sims about the use of artificial intelligence in predicting response to breast cancer treatment on CTV on Friday September 25 , 2015: Link