

Tag Archives: information theory
Sept. 7, 2019. Pan cancer article and database update
We have published another revision to our pan-cancer splicing mutation database paper: Shirley BC, Mucaki EJ and Rogan PK. Pan-cancer repository of validated natural and cryptic mRNA splicing mutations F1000Research 2019, 7:1908 (https://f1000research.
In this version, we derive a simplified variant classification scheme with ClinVar designations calibrated to the molecular phenotypes of mRNA splicing mutations. These are now indicated in the ValidspliceMut (https://validsplicemut.
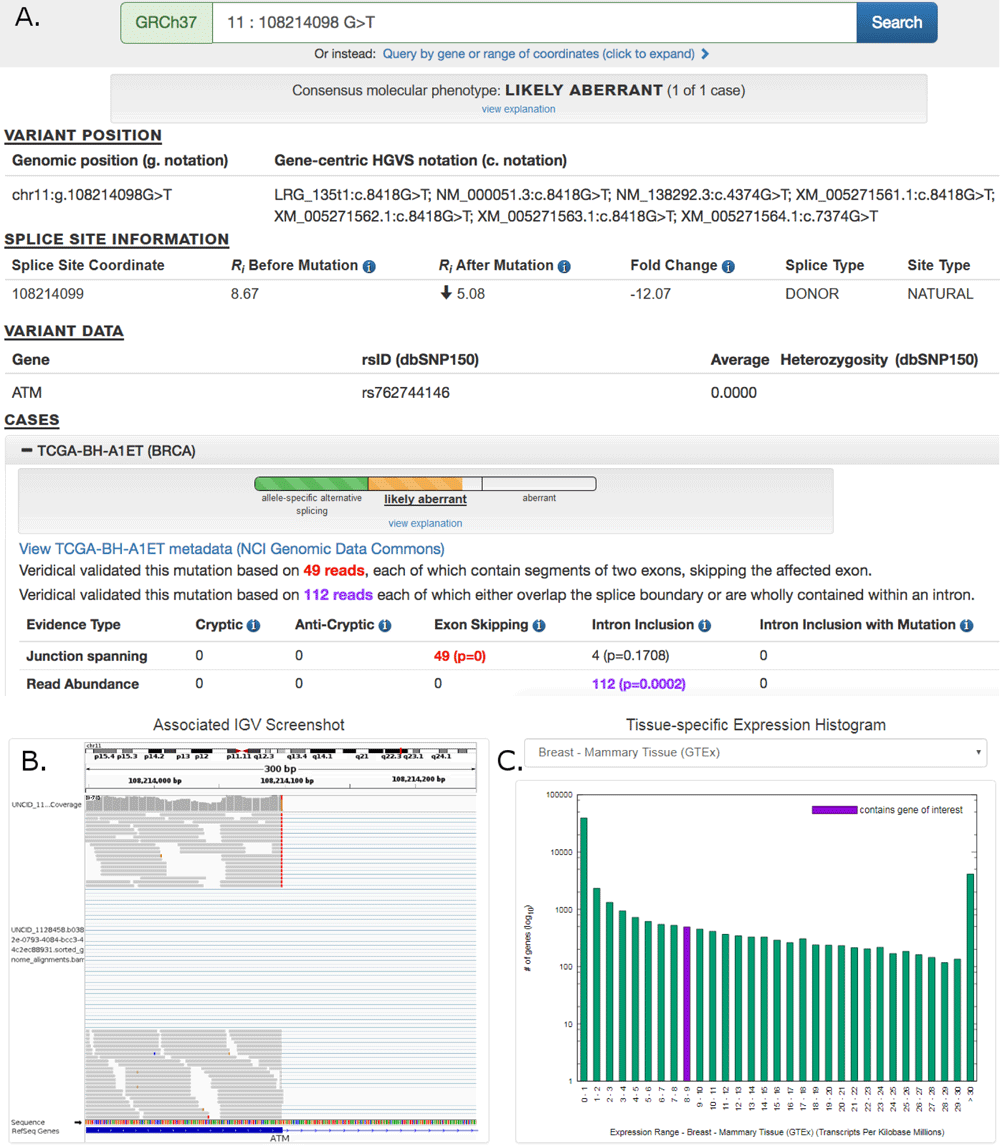
Figure 1 of Pan-cancer repository of validated natural and cryptic mRNA splicing mutations. F1000Research 2019, 7:1908
March 20, 2019. Presentation at the 2019 American College of Medical Genetics and Genomics annual conference
The following paper has been accepted for presentation:
“Pan-Cancer Repository of Validated Natural and Cryptic mRNA Splicing Mutations”,
Category: “Laboratory genetics and genomics”, Abstract Poster Number: 754 (link to Abstract)
Where: Exhibit hall, Washington Convention Center, ACMG Clinical Genetics Meeting in Seattle, Washington
When: April 2 – 6, 2019; Poster presentation time: Friday, 4/5 from 10:30am-12:00pm
This work will be available as an ePoster AS WELL AS being presented in printed format on a poster board during the Annual Meeting. Details to access the ePoster will be available soon.
March 17, 2015. Review article on mRNA splicing mutations highlighted on F1000Research blog
Revised version of our review article on mRNA splicing in F1000Research has been highlighted in the Faculty of 1000 blog:
http://blog.f1000research.com/2015/03/17/interpreting-genomic-variants-in-rare-and-common-diseases/
The blog entry includes links to our YouTube video:
Short version (3:42)
Long version (9:41)
July 13, 2014. Presentations at the 2014 American Society of Human Genetics Conference
Cytognomix will be presenting several papers at the upcoming ASHG annual meeting (October 18-22, 2014, San Diego):
Using information theory to analyze and predict splicing mutations in rare and common diseases: performance and best practices. N.G. Caminsky, E. Mucaki and P.K. Rogan
Reversing differences in chromatin accessibility that distinguish homologous mitotic metaphase chromosomes. W.A. Khan, P.K. Rogan, J.H.M. Knoll
Automated Dicentric Chromosome Identification by Machine Learning-based Image Processing. P.K. Rogan, Y. Li, A. Subasinghe, J. Samarabandu, R. Wilkins and J.H. Knoll
Towards the minimal breast cancer genome and its relevance to chemotherapy. S.N. Dorman, J.H. Knoll, K. Baranova, C. Viner, P.K. Rogan
The FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping and is a risk factor for familial breast cancer. Paolo Peterlongo , Francesca Damiola, Eliseos Mucaki, Valentina Dall’Olio ,Sara Pizzamiglio , Irene Catucci , Anders Kvist , Paolo Verderio, Mara Colombo , Loris Bernard , Hans Ehrencrona, Laura Caleca, Valeria Pensotti , Sylvie Mazoyer, Peter K. Rogan ,Paolo Radice
Please contact us if you would like to meet or discuss this work.
January 4, 2013. Paper: “Predicting mRNA transcript isoforms derived from splicing mutations”, ASSEDA server

Volume 34, Issue 4
“Prediction of mutant mRNA splice isoforms by information theory-based exon definition,” by Eliseos Mucaki, Ben Shirley and Peter Rogan has been accepted for publication by the journal Human Mutation.
Abstract. Mutations that affect mRNA splicing often produce multiple mRNA isoforms, resulting in complex molecular phenotypes. Definition of an exon and its inclusion in mature mRNA relies on joint recognition of both acceptor and donor splice sites. This study predicts cryptic and exon skipping isoforms in mRNA produced by splicing mutations from the combined information contents (Ri, which measures binding site affinity) and distribution of the splice sites defining these exons. The total information content of an exon (Ri,total) is the sum of the Ri values of its acceptor and donor splice sites, adjusted for the distance separating these sites, ie. the gap surprisal. Differences between total exon information contents (ΔRi,total) are predictive of the relative abundance of these exons in distinct processed mRNAs. Constraints on splice site and exon selection are used to eliminate non-conforming and poorly expressed isoforms. Molecular phenotypes are computed by the Automated Splice Site and Exon Definition Analysis server (ASSEDA; http://splice.uwo.ca). Predictions of splicing mutations were highly concordant (85.2%; n=61) with published expression data. In silico exon definition analysis will contribute to streamlining assessment of abnormal and normal splice isoforms resulting from mutations.
Update: The paper is now available online from the Journal website: DOI: 10.1002/humu.22277 and is cited on PubMed.
Update 2: John Mucaki has produced a Video Tutorial on using the ASSEDA server on YouTube.
Update 3: The accepted paper has now been copyedited, typeset and published online: http://onlinelibrary.wiley.com/doi/10.1002/humu.22277/abstract. Supplementary data are available as well. (2-21-2013)
Update 4: Annual subscriptions to the Automated Splice Site and Exon Definition server are available through Cytognomix (2-22-2013).
Update 5: The paper has been highlighted in the April 2013 issue of the Journal, where it appeared. Bing Yu, University of Sydney, authored the commentary (Vol 34[4], page v).
Update 6: Mucaki EJ., Shirley BC, and Rogan PK. Prediction of Mutant mRNA Splice Isoforms by Information Theory-Based Exon Definition has been published in print. Human Mutation, April 2013, Volume 34 (4), pages 557–565. The journal has made the paper FREE for anyone to download.
Resources
Links to the latest CytoGnomix products:
Applications and consulting in Geostatistical Epidemiology
Monitoring and discriminating infectious disease hotspots from high disease burden regions, eg. for COVID-19:
Zenodo repository: Geostatistical Analysis of SARS-CoV-2 Positive Cases in the United States
Defence Canada IDEaS project: Locating emerging COVID19 hotspots in Ontario after community transmission by time-correlated, geospatial analysis
Addressing large scale radiation incidents and accidents:
Article in PLOS One: Meeting radiation dosimetry capacity requirements of population-scale exposures …. (Funded by High performance computing consortium: SOSCIP and CytoGnomix)
How to: Protocol for Geostatistical Determination of Radiation Dosimetry Maps of Population-Scale Exposures
Large scale Radiation Biodosimetry
Capacity of supercomputer version of Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) software: Automated Cytogenetic Biodosimetry at Population-Scale and Radiation, Radiation, 2021 (link to published article).
Scalable, democratized access to ADCI:
Overview of Cloud version- ADCI_Online
Presentation to the International Atomic Energy Agency (CRP E35010)
Gene Expression Signatures for Radiation Biodosimetry
Mucaki, E.J., Shirley, B.C. and Rogan, P.K., 2021. Improved radiation expression profiling in blood by sequential application of sensitive and specific gene signatures. International Journal of Radiation Biology, doi.org/10.1080/09553002.2021.1998709 Link to pdf: Improved radiation expression profiling…
Zhao, J.Z., Mucaki, E.J. and Rogan, P.K., 2018. Predicting ionizing radiation exposure using biochemically-inspired genomic machine learning. F1000Research, 7(233), p.233. Link to open access article: https://f1000research.com/articles/7-233
Large Scale Repository of Cancer Splicing Mutations
Pan-cancer repository of validated natural and cryptic mRNA splicing mutations (a major public resource of mRNA splicing mutations validated according to multiple lines of evidence of abnormal gene expression. )
Article in F1000Research: Pan-Cancer repository of …..
Presentation at the 2019 American College of Medical Genetics Annual Meeting:
Pan-cancer repository of validated natural and cryptic mRNA.ePoster
Interactive Website: Gene signatures for chemotherapy drug response
Demo (Windows): Automated Dicentric Chromosome Identifier and Dose Estimator
Review on information theory-based splicing mutation analysis:
Caminsky et al. 2014, Videos describing this paper: short and long versions.
Mission
We find and validate mutations that others cannot…
with advanced, patented genomic probe and bioinformatic technologies.
CytoGnomix continues our long track record of creating technologies for genomic medicine.
We anticipate and implement the needs of the biomedical and clinical genomics communities.