Volume 34, Issue 4
“Prediction of mutant mRNA splice isoforms by information theory-based exon definition,” by Eliseos Mucaki, Ben Shirley and Peter Rogan has been accepted for publication by the journal Human Mutation.
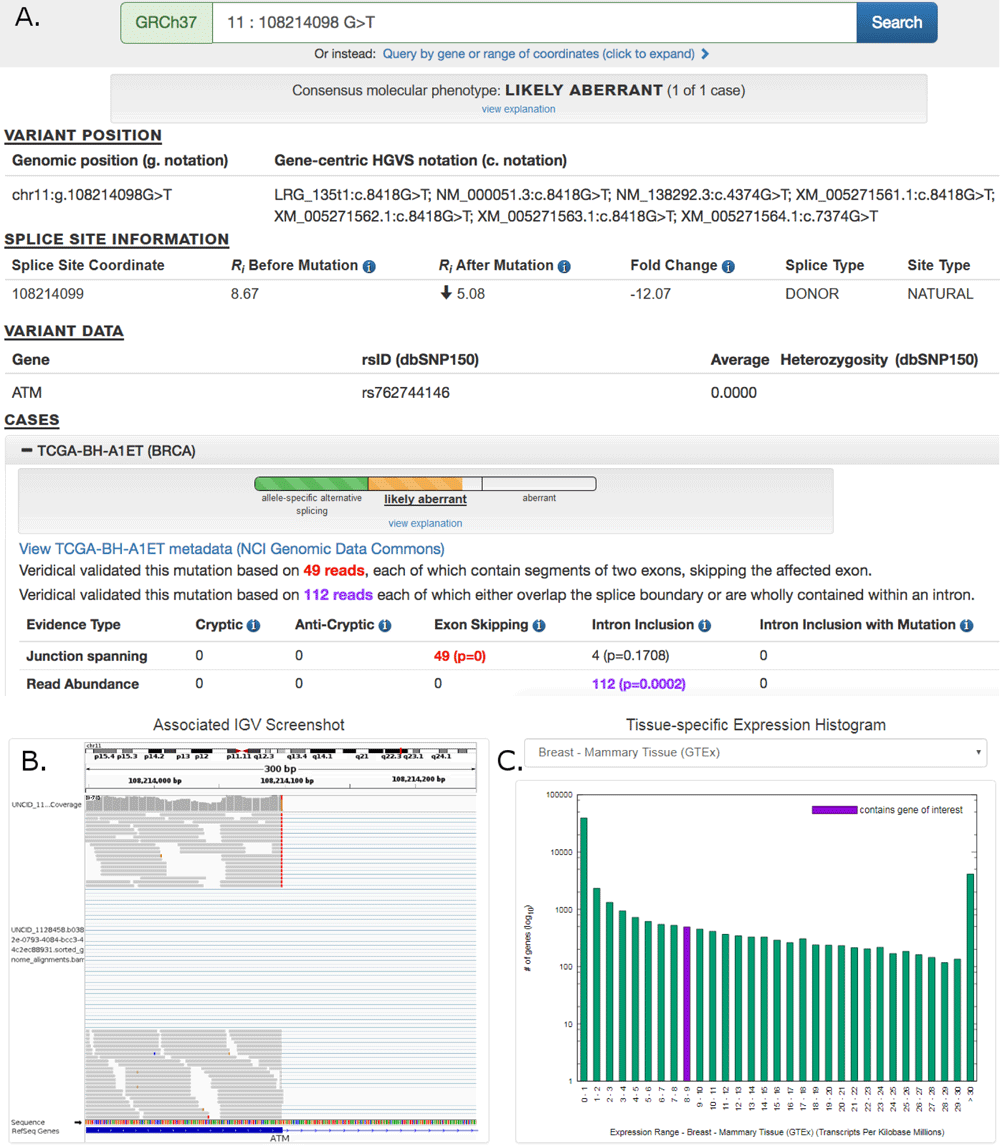
Abstract. Mutations that affect mRNA splicing often produce multiple mRNA isoforms, resulting in complex molecular phenotypes. Definition of an exon and its inclusion in mature mRNA relies on joint recognition of both acceptor and donor splice sites. This study predicts cryptic and exon skipping isoforms in mRNA produced by splicing mutations from the combined information contents (Ri, which measures binding site affinity) and distribution of the splice sites defining these exons. The total information content of an exon (Ri,total) is the sum of the Ri values of its acceptor and donor splice sites, adjusted for the distance separating these sites, ie. the gap surprisal. Differences between total exon information contents (ΔRi,total) are predictive of the relative abundance of these exons in distinct processed mRNAs. Constraints on splice site and exon selection are used to eliminate non-conforming and poorly expressed isoforms. Molecular phenotypes are computed by the Automated Splice Site and Exon Definition Analysis server (ASSEDA; http://splice.uwo.ca). Predictions of splicing mutations were highly concordant (85.2%; n=61) with published expression data. In silico exon definition analysis will contribute to streamlining assessment of abnormal and normal splice isoforms resulting from mutations.
Update: The paper is now available online from the Journal website: DOI: 10.1002/humu.22277 and is cited on PubMed.
Update 2: John Mucaki has produced a Video Tutorial on using the ASSEDA server on YouTube.
Update 3: The accepted paper has now been copyedited, typeset and published online: http://onlinelibrary.wiley.com/doi/10.1002/humu.22277/abstract. Supplementary data are available as well. (2-21-2013)
Update 4: Annual subscriptions to the Automated Splice Site and Exon Definition server are available through Cytognomix (2-22-2013).
Update 5: The paper has been highlighted in the April 2013 issue of the Journal, where it appeared. Bing Yu, University of Sydney, authored the commentary (Vol 34[4], page v).
Update 6: Mucaki EJ., Shirley BC, and Rogan PK. Prediction of Mutant mRNA Splice Isoforms by Information Theory-Based Exon Definition has been published in print. Human Mutation, April 2013, Volume 34 (4), pages 557–565. The journal has made the paper FREE for anyone to download.