
 Our patent application, US Patent Application Serial Number 17/137,317, has had all claims allowed by the US Patent and Trademark office. This application covers the method underlying our ADCI Radiation biodosimetry software system. New claims cover partial body exposures, which are typical in radiation therapy. In addition, this invention covers applications of the technology which do not require interaction with the microscope system software, and can be used as a standalone system. The patent should be issued within the next several months.
Our patent application, US Patent Application Serial Number 17/137,317, has had all claims allowed by the US Patent and Trademark office. This application covers the method underlying our ADCI Radiation biodosimetry software system. New claims cover partial body exposures, which are typical in radiation therapy. In addition, this invention covers applications of the technology which do not require interaction with the microscope system software, and can be used as a standalone system. The patent should be issued within the next several months.
Tag Archives: cytogenetic
October 20, 2021. New article on metaphase epigenetics published in Molecular Cytogenetics journal
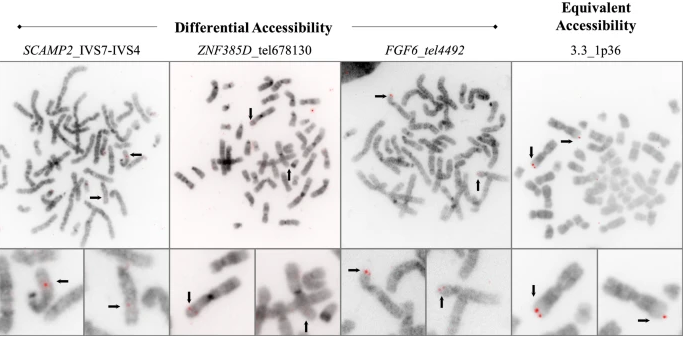
Hill, S.L., Rogan, P.K., Wang, Y.X. Knoll J.H.M. Differentially accessible, single copy sequences form contiguous domains along metaphase chromosomes that are conserved among multiple tissues. Mol Cytogenet 14, 49 (2021). https://molecularcytogenetics.biomedcentral.com/articles/10.1186/s13039-021-00567-w
March 29, 2021. Article describing accelerated biodosimetry by high performance computing
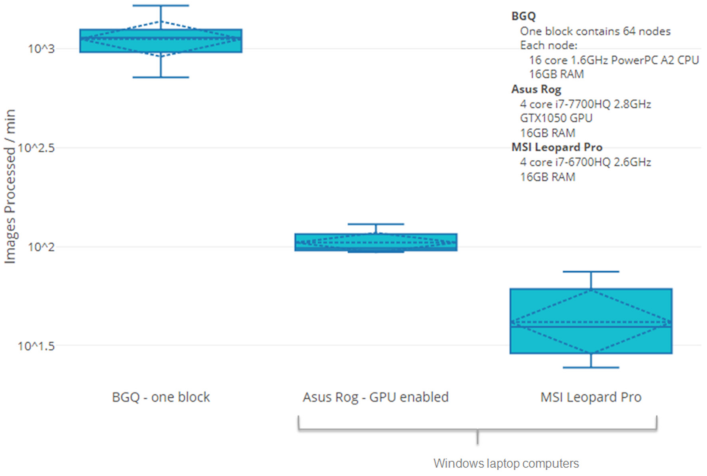
We have published a new article about accelerating biodosimetry testing in a large scale radiation incident:
Rogan PK, Mucaki EJ, Shirley BC, Li Y, Wilkins RC, Norton F, Sevriukova O, Pham N-D, Waller E, Knoll JHM. Automated Cytogenetic Biodosimetry at Population-Scale. Radiation. 2021; 1(2):79-94. doi: 10.3390/radiation1020008 (2021)
Nov. 9, 2020. Notice of Allowance for US Pat App. Ser. No. 16/057,710
CytoGnomix’s Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) system will be awarded a US Patent for all claims covering “Smart Microscope System for Radiation Biodosimetry.” The patent application is available at:
https://patents.google.com/patent/US20200050831A1
The abstract reads:
An automated microscope system is described that detects dicentric chromosomes (DCs) in metaphase cells arising from exposure to ionizing radiation. The radiation dose depends on the accuracy of DC detection. Accuracy is increased using image segmentation methods are used to rank high quality cytogenetic images and eliminate suboptimal metaphase cell data in a sample based on novel quality measures. When a sufficient number of high quality images are detected, the microscope system is directed to terminate metaphase image collection for a sample. The microscope system integrates image selection procedures that control an automated digitally controlled microscope with the analysis of acquired metaphase cell images to accurately determine radiation dose. Early termination of image acquisition reduces sample processing time without compromising accuracy. This approach constitutes a reliable and scalable solution that will be essential for analysis of large numbers of potentially exposed individuals.
September 4, 2020. New article on automated partial body radiation exposure determination
We have added the capability to determine whether samples exposed to ionizing radiation are wholly or partially irradiated. If partially, the approach determines the fraction of metaphase cells exposed and the whole body-equivalent dose completely automatically. CytoGnomix’s Automated Dicentric Chromosome Identifier and Dose Estimation software has been upgraded to generate these results as part of the the dose estimation report. The article has been accepted for publication by the International Journal of Radiation Biology V. 96 (https://doi.org/10.1080/09553002.2020.1820611). It is also currently available on BioRxiv:
Estimating partial body ionizing radiation exposure by automated cytogenetic biodosimetry
July 4, 2019. Presentations describing interlaboratory comparison of radiation exposure determination by automated cytogenetic biodosimetry
We will be presenting:
Determination of radiation exposure levels by fully automated
dicentric chromosome analysis: Results from IAEA MEDBIODOSE
(CRP E35010) interlaboratory comparison
at both the 19th International Congress of Radiation Research (Aug. 25-29, 2019) and the 12th International Symposium on Chromosome Aberrations (Aug. 27, 2019) in Manchester, UK. This study compared the performance of our Automated Dicentric Chromosome Identifier and Dose Estimator (ADCI) using data from 6 different laboratories. Each of these members of the IAEA-sponsored Cooperative Research Project E35010, submitted images for calibration curve construction and at least 2 samples of unknown exposure to CytoGnomix for analysis with ADCI. We will report the results of this analysis during this presentation.
This poster presentation is now available on the Zenodo website (http://doi.org/10.5281/zenodo.4012749)
doi: DOI 10.5281/zenodo.4012748
Authors:
Rogan P , Shirley B , Li Y , Guogyte K , Sevriukova O , Ngoc Duy P , Moquet J ,
Ainsbury E , Sudprasert W , Wilkins R , Norton F , Knoll J
Department of Biochemistry , University of Western Ontario, London Ontario, Canada
Department of Pathology and Laboratory Medicine, University of Western Ontario, London
Ontario, Canada
Radiation Protection Centre, Ministry of Health (L T -RPC), Vilnius, Lithuania
Dalat Nuclear Research Institute (VN-DNRI), Dalat, Vietnam
Public Health England (PHE), Oxford, Great Britain
Thai Biodosimetry Network, Kasetsart University (THA), Bangkok, Thailand
Health Canada, Ottawa Ontario, Canada
Canadian Nuclear Laboratories, Chalk River Ontario, Canada
Cytognomix, London Ontario, Canada
January 9, 2019. Article on fully automated interpretation of the dicentric chromosome assay for radiation quantification now available
Our paper, “RADIATION DOSE ESTIMATION BY COMPLETELY AUTOMATED INTERPRETATION OF THE DICENTRIC CHROMOSOME ASSAY” is now published in the journal Radiation Protection Dosimetry.
Unfortunately, the journal has not made the article open access. We have made it available on our ADCIWiki website, as permitted by the copyright agreement.
The link to the pdf full text is:
April 22, 2014. New paper describing Automated Biodosimetry Software published
We have published an article in Radiation Protection Biodosimetry describing our patented Automated Dicentric Chromosome Identifier Software for both Desktop and Supercomputer systems. The citation is:
Peter K. Rogan, Yanxin Li, Asanka Wickramasinghe, Akila Subasinghe, Natasha Caminsky, Wahab Khan, Jagath Samarabandu, Ruth Wilkins, Farrah Flegal, and Joan H. Knoll. AUTOMATING DICENTRIC CHROMOSOME DETECTION FROM CYTOGENETIC BIODOSIMETRY DATA. Radiat Prot Dosimetry. first published online April 21, 2014 doi:10.1093/rpd/ncu133 (Rogan et al. Radiat Prot Dosimetry. 2014).
This paper was presented at the EPR Biodose 2013 meeting in Leiden, Netherlands. The software identifies highly variable features in a large quantity images in relatively short time frame. Multiple technologies are employed, including SVM machine learning, gradient vector flow, parallelization, and other methods.
September 19. Abstract on metaphase epigenetics: platform presentation at American Society of Human Genetics meeting
Non-random, locus-specific differences in DNA accessibility are present in homologous metaphase chromosomes. W. A. Khan1,3, P. K. Rogan2,3,4, J. H. M. Knoll1,3,4 1) Department of Pathology; 2) Departments of Biochemistry and Computer Science; 3) University of Western Ontario, London, Ontario, Canada; 4) Cytognomix, London, Ontario, Canada.
/
Condensation differences between heterochromatin and euchromatin along the lengths of homologous, mitotic metaphase chromosomes are well known. This study describes differences in metaphase compaction between homologous euchromatic loci. We report molecular cytogenetic data showing local differences in condensation between homologs that are related to differences in accessibility (DA) of associated DNA probe targets. Reproducible DA was observed at ~10% of 450 distinct genomic regions mapped by single copy fluorescence in situ hybridization (scFISH). Fourteen short (1.5-5kb) sc and low copy (lc) FISH probes (from chromosomes 1, 5, 9, 11, 15, 16, 17, 22) targeting genic and non-genic regions with and without DA were developed and hybridized to cells from 10 individuals with cytogenetically-distinguishable homologs. Differences in hybridization were non-random for 6 genomic regions (RGS7, CACNAB1, HERC2, PMP22:IVS3, ADORA2B:IVS1, ACR) and were significantly-biased towards the same homolog (p< 0.01; n = 355 cells). The imprinted paternal chromosome 15 in a three-generation pedigree also showed non-random bias in DA. DNA probes within CCNB1, C9orf66, ADORA2B:Ex 1-IVS1, PMP22:IVS4-Ex 5, and a nongenic region within 1p36.3 did not show DA, while OPCML showed unbiased DA. A subset of probes was mapped onto chromosome topography by FISH-correlated atomic force microscopy (AFM). To quantify DA and pinpoint probe locations, we performed 3D-structured illumination super-resolution microscopy (3D-SIM). 3D anaglyph videos showed genomic regions with DA having nearly 5-fold larger differences in volumetric integrated probe intensities between homologs. Additional non-DA probes (NOMO1, NOMO3) hybridized to grooves in chromosome topography and exhibited a narrow range of probe depths (average: 0.08 μm) along axial and lateral axes of the 2 homologs. In contrast, probe for targets with DA (HERC2, PMP22:IVS3, ACR) significantly differed in probe depth (average: 0.77 μm) and volume (p < 0.05) between each homolog. Interestingly, genomic regions without DA are enriched in epigenetic marks (DHS, H3K27Ac, H3K4me1) of accessible interphase chromatin to a greater extent than regions with DA, suggesting these differences may be correlated with epigenetic marks established during the previous interphase. In summary, we present several lines of evidence that regional differences in condensation between homologs are programmed during metaphase chromosome compaction.
Click here for presentation details: session, location, time.
January 28, 2013. Platform presentation on Automated Dicentric Chromosome Identifier Software
“Automating Dicentric Chromosome Detection from Cytogenetic Biodosimetry Data” at the International EPRBioDose 2013 Conference in Leiden, Netherlands (March 24-28).
Authors: Peter Rogan(1,2), Akila Subasinghe(1), Asanka Wickramasinghe(1), Yanxin Li(1), Jagath Samarabandu(1), Joan Knoll(1,2), Ruth Wilkins(3), Farah Flegal(4); (1)University of Western Ontario, (2)Cytognomix Inc., (3)Health Canada, (4)Atomic Energy of Canada Ltd., Canada.
Abstract: We are developing a prototype software system with sufficient capacity and speed to estimate radiation exposures by counting dicentric chromosomes in metaphase cells from many individuals in the event of a
mass casualty. Top-ranked metaphase images are segmented by defining chromosomes with an active contour gradient vector field (GVF), and by determining centromere locations along the centerline. The centerline is
extracted by Discrete Curve Evolution (DCE) skeleton branch pruning and curve interpolation. Centromere detection minimizes the global width and DAPI-staining intensity profiles along the centerline. A second
centromere is identified by reapplying this procedure after masking the first. Dicentrics can be identified by applying a support vector machine-based classification, which uses features that capture width and intensity
profile characteristics as well as local shape features of the object contour at candidate pixel locations. The correct location of the centromere is also refined in chromosomes with sister chromatid separation. The
overall algorithm has both high sensitivity (85%) and specificity (94%). Results are independent of the shape and structure of chromosomes in different cells, regardless of which laboratory protocol is followed or the
specimen source. The requisite throughput is being achieved by recoding MATLAB software modules for different segmentation functions in C++/OpenCV, and integrating them in the prototype. Processing of
numerous images is accelerated by both data and task software parallelization with the Message Passaging Interface and Intel Threading Building Blocks as well as an asynchronous non-blocking I/O strategy. Relative
to a serial process, metaphase ranking, GVF, and DCE are respectively 100 and 300 fold faster on an 8-core I7-based desktop and on a 64-core shared memory cluster computer. Extrapolation from these benchmarks to
a 64-core system in which all of the software modules have been integrated indicates that it should be feasible to process metaphases for dicentric chromosomes from 1000 specimens in 20 hours.