

Author Archives: Peter Rogan
Sept. 18, 2015. Press release about chemotherapy resistance paper
Western University hopes to use artificial intelligence to improve breast cancer patient outcomes.
(http://mediarelations.uwo.ca/2015/09/18/researchers-at-western-university-hope-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes/, other links at end of post)
Western University researchers are working on a way to use artificial intelligence to predict a patient’s response to two common chemotherapy medications used to treat breast cancer – paclitaxel and gemcitabine.
Peter Rogan, PhD, and a team of researchers, including Stephanie Dorman, PhD, and Katherina Baranova, BMSc, at Western’s Schulich School of Medicine & Dentistry, are hoping to one day remove the guesswork from breast cancer treatment with this technique.
Based on personal genetic analysis of their tumours, patients with the same type of cancer can have different responses to the same medication. While some patients will respond well and go into remission, others will develop a resistance to the medication.
Identifying the genetic factors which lead to resistance or remission can help develop better targeted, individualized treatment regimens with better patient outcomes.
“Treating patients with therapies that are the most likely to be successful can help reduce unnecessary toxicity and improve overall outcomes,” said Dorman.
Rogan and Joan Knoll, PhD, professor, Schulich Medicine & Dentistry, began by defining a stable set of genes in 90 per cent of breast cancer tumours in 2012.
Beginning with 40 genes including several stable genes, the team then used artificial intelligence combined with data from cell lines and tumour tissue from cancer patients who had treatment with at least one of the medications to narrow down and identify the genetic signatures most important for determining resistance and remission for each medication. Their study has recently been published in the journal, Molecular Oncology.
Using the data, the researchers were able to identify the 84 per cent of women with breast cancer who would go into remission in response to the drug paclitaxel. The genetic signature identified for the drug gemcitabine was able to predict remission using preserved tumour tissue with 62 to 71 per cent accuracy.
Now, with this data in hand, the researchers are working to further refine the genetic signatures and improve the predictions further.
“Artificial intelligence is a powerful tool for predicting drug outcomes because it looks at the sum of all the interacting genes,” said Rogan, professor in the departments of Biochemistry, Oncology and Computer Science, Canada Research Chair in Genome Bioinformatics and president, Cytognomix Inc. “If we can use this technology to improve our knowledge of which medications to use, it could improve patient outcomes. The earlier we treat a patient with the most effective medication, the more likely we can effectively treat or possibly even cure that patient.”
###
Reference: Dorman SN, Baranova K, Knoll JH, Urquhart BL, Mariani G, Carcangiu ML, Rogan PK. Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Mol Oncol. 2015 Aug 22. pii: S1574-7891(15)00146-5. doi: 10.1016/j.molonc.2015.07.006. [Epub ahead of print] http://www.moloncol.org/article/S1574-7891%2815%2900146-5/fulltext
MEDIA CONTACT: Tristan Joseph, Media Relations Officer, Schulich School of Medicine & Dentistry, Western University, 519-661-2111 ext. 80387, c: 519-777-1573, tristan.joseph@schulich.uwo.ca
ABOUT WESTERN
Western delivers an academic experience second to none. Since 1878, The Western Experience has combined academic excellence with life-long opportunities for intellectual, social and cultural growth in order to better serve our communities. Our research excellence expands knowledge and drives discovery with real-world application. Western attracts individuals with a broad worldview, seeking to study, influence and lead in the international community.
ABOUT THE SCHULICH SCHOOL OF MEDICINE & DENTISTRY
The Schulich School of Medicine & Dentistry at Western University is one of Canada’s preeminent medical and dental schools. Established in 1881, it was one of the founding schools of Western University and is known for being the birthplace of family medicine in Canada. For more than 130 years, the School has demonstrated a commitment to academic excellence and a passion for scientific discovery.
Follow Western Media Relations online:
Website: http://communications.uwo.ca/media/
RSS: http://feeds.feedburner.com/MediaWesternU
Twitter: https://twitter.com/mediawesternu
Links to story:
(http://www.eurekalert.org/pub_releases/2015-09/uowo-wuh091815.php,
http://www.1069thex.com/2015/
http://medicalxpress.com/news/2015-09-artificial-intelligence-breast-cancer-patient.html,
http://u-s.news/researchers-hope-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes,
http://linkis.com/eurekalert.org/Western_University_h.html,
http://jerseytribune.com/2015/09/18/western-university-hopes-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes/,
http://www.dotmed.com/news/story/27121,
http://www.sciencenewsline.com/summary/2015091817530026.html,
http://www.demanjo.com/news/health/5084082/western-university-hopes-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes.html,
http://danhartshorn.com/2015/09/researchers-hope-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes/,
http://www.bioportfolio.com/news/article/2463671/Western-University-hopes-to-use-artificial-intelligence-to-improve-breast-cancer-patient.html,
http://scifeeds.com/news/western-university-hopes-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes/,
http://www.biospace.com/news_story.aspx?StoryID=391867&full=1,
http://breastcancer.einnews.com/article__detail/287108153-western-university-hopes-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes?vcode=XIbw,
https://twitter.com/cambridgepharma,
http://www.newslocker.com/en-uk/news/science/western-university-hopes-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes/,
http://gadgets.ndtv.com/science/news/artificial-intelligence-could-improve-breast-cancer-treatment-742015,
http://www.news-medical.net/news/20150919/Artificial-intelligence-may-improve-outcomes-in-breast-cancer-patients.aspx,
http://odishasamaya.com/news/artificial-intelligence-may-be-the-antidote-to-breast-cancer/55895,
http://focusnews.com/lifestyle/artificial-intelligence-could-improve-breast-cancer-treatment/127603/,
http://www.thehansindia.com/posts/index/2015-09-20/Artificial-intelligence-could-improve-breast-cancer-treatment-176708,
http://www.freepressjournal.in/artificial-intelligence-could-improve-breast-cancer-treatment/,
http://www.thehealthsite.com/news/a-new-way-to-gauge-breast-cancer-drug-efficacy-on-the-cards/,
http://www.dailypioneer.com/health-and-fitness/artificial-intelligence-could-improve-breast-cancer-treatment.html,
http://timesofindia.indiatimes.com/life-style/health-fitness/health-news/Artificial-intelligence-could-improve-breast-cancer-treatment/articleshow/49024103.cms,
http://zeenews.india.com/news/health/health-news/artificial-intelligence-could-improve-breast-cancer-treatment_1799356.html,
http://www.canindia.com/2015/09/artificial-intelligence-could-improve-breast-cancer-treatment/,
http://www.daijiworld.com/news/news_disp.asp?n_id=355231,
http://www.business-standard.com/article/news-ians/artificial-intelligence-could-improve-breast-cancer-treatment-115091900215_1.html,
http://www.newkerala.com/news/2015/fullnews-122242.html,
http://www.healthcanal.com/cancers/breast-cancer/67107-researchers-at-western-university-hope-to-use-artificial-intelligence-to-improve-breast-cancer-patient-outcomes.html,
http://www.exchangemagazine.com/morningpost/2015/week38/Monday/15092106.htm,
https://www.facebook.com/breastcancersocietyofcanada?fref=nf,
http://www.medicaldaily.com/artificial-intelligence-may-be-able-predict-remission-or-resistance-certain-drugs-353628,
http://www.gizmag.com/machine-learning-predicts-breast-cancer-treatment-responses/39510/,
http://www.sciencecodex.com/artificial_intelligence_to_improve_breast_cancer_patient_outcomes-165300,
http://www.dqindia.com/can-artificial-intelligence-be-used-to-improve-breast-cancer-treatment/,
http://www.science20.com/news_articles/artificial_intelligence_to_improve_breast_cancer_patient_outcomes-157221,
http://geekview.baijia.baidu.com/article/179752,
http://md.tech-ex.com/2015/news/oversea/41383.html,
http://www.cnbeta.com/articles/432439.htm)
September 11, 2015. Final version of paclitaxel and gemcitabine chemotherapy signature paper now published

Stephanie N. Dorman, Katherina Baranova, Joan H.M. Knoll, Brad L. Urquhart, Gabriella Mariani, Maria Luisa Carcangiu, Peter K. Rogan
Received: July 20, 2015; Accepted: July 31, 2015; Published Online: August 21, 2015
Publication stage: In Press, Corrected Proof
September 7, 2015. Video presentation of Molecular Oncology article on chemotherapy response
We have published a video synopsis of :
Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Stephanie N. Dorman, Katherina Baranova, Joan H.M. Knoll, Brad L. Urquhart, Gabriella Mariani, Maria Luisa Carcangiu, Peter K. Rogan. Molecular Oncology, in press. DOI: http://dx.doi.org/10.1016/j.molonc.2015.07.006
August 28, 2015. Article on chemotherapy gene signature published
The uncorrected proofs of our new paper:
Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Dorman et al. Mol. Oncol. 2015
DOI: 10.1016/j.molonc.2015.07.006
are now available online at this link (http://dx.doi.org/10.1016/j.
Aug. 25, 2015. Top viewed article in Molecular Cytogenetics
Our article:
Reversing chromatin accessibility differences that distinguish homologous mitotic metaphase chromosomes. Wahab Khan, Peter Rogan, Joan Knoll. Molecular Cytogenetics 2015, 8:65
was published on August 13th. In less than two weeks, it has become the most viewed article in this journal for the past month, averaging 55 per day.
Update: as of Sept. 6, the article is still the top viewed paper with 887 views.
August 13, 2015. New paper on metaphase epigenetics published
The next exciting installment of the “story” about differential accessibility of metaphase chromosomes has been published. Cytognomix’s single copy FISH technology was key to making these observations.
Reversing chromatin accessibility differences that distinguish homologous mitotic metaphase chromosomes. Khan et. al. Molecular Cytogenetics 2015, 8:65
July 31, 2015. Chemotherapy resistance in breast cancer manuscript accepted for publication
Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning
Authors: Stephanie N. Dormana, Katherina Baranovaa, Joan H.M. Knollb,c,d, Brad L. Urquharte, Gabriella Marianif, Maria Luisa Carcangiuf, Peter K. Rogana,d,g,h*
aDepartment of Biochemistry, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada, bDepartment of Pathology and Laboratory Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada, cMolecular Diagnostics Division, Laboratory Medicine Program, London Health Sciences Centre, ON, Canada, dCytognomix Inc., London ON, Canada, eDepartment of Physiology and Pharmacology, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada, fDepartment of Diagnostic and Laboratory Pathology, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy, gDepartment of Computer Science, University of Western Ontario, London, ON, Canada, hDepartment of Oncology, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada
will be published in a forthcoming issue of the journal “Molecular Oncology”.
July 11, 2015. Presentation at RegGen Satellite Meeting at 2015 ISMB Annual Meeting
We presented:
“Discovery of Primary, Cofactor, and Novel Transcription Factor Binding Site Motifs by Recursive, Thresholded Entropy Minimization”
by Ruipeng Lu, Eliseos Mucaki, and Peter Rogan
at the Regulatory Genomics Special Interest Group meeting in Dublin, Ireland: Link to abstract
July 8, 2015. New paper using Cytognomix’s single copy FISH probes
Khan WA, Rogan PK, Knoll JH. Reversing chromatin accessibility differences that distinguish homologous mitotic metaphase chromosomes. Molecular Cytogenetics, in press.
Stay tuned for posts providing details and links to the manuscript once it is available online at the journal website.
July 3, 2015. New publication on breast cancer gene mutation
FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity, and is a familial breast cancer risk factor.
Peterlongo et al. Hum Mol Genet. 2015 Jun 30. pii: ddv251.
In this paper, we use information theory to demonstrate a new mechanism for disease mutations. It turns out that this a fairly common type of mutation in our unpublished studies of other data sources.
June 27, 2015. Best oral presentation at the 12th Annual London Oncology Research & Education Conference
Natasha Caminsky presented:
A Unified Framework for the Identification and Prioritization of Coding and Non-Coding Variants in Heritable Breast and Ovarian Cancer (HBOC).
which introduces Cytognomix’s approach for analysis of a wide range of regulatory mutations in complete human gene and genome data. The other authors of this study were Mucaki EJ, Lu R, Perri AM, and Rogan PK.
This is an annual event for scientists, clinicians, graduate students and Post-Doctoral fellows at the University of Western Ontario and affiliated hospitals to share cancer research discoveries and promote cancer research collaboration and training. She was awarded Best Oral Presentation for this paper.
The Breast Cancer Society of Canada blogged the results of the paper competition: link
April 18, 2015. New software distribution agreement for MutationForecaster
Today, Cytognomix Inc. and Illumina signed a distribution agreement to make MutationForecaster software available through the BaseSpace ecosystem. Work is underway to enable Illumina users to analyze data processed in BaseSpace to be interpreted with Cytognomix’s software. The BaseSpace environment enables MiSeq users to carry out sequence analyses with requiring an onsite computing infrastructure, with scalable cloud data storage, to manage all of your data analyses, to securely collaborate, and to share and access Illumina and community-developed applications. MutationForecaster will import variant control format files, validate them with RNASeq Bam files from the same sample directly from BaseSpace and download results from Cytognomix back to BaseSpace.
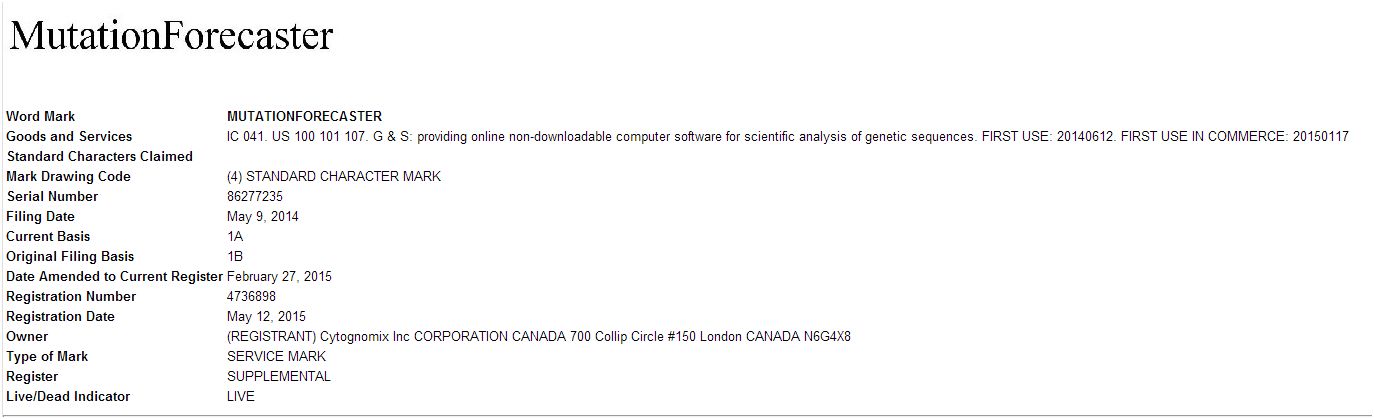
May 22, 2015. MutationForecaster receives US Trademark
On May 12, the US Patent and Trademark Office awarded CytoGnomix a trademark for:
MutationForecaster®
May 21, 2015. Platform presentation at Compute Ontario Research Day
Dr. Peter Rogan’s laboratory at the University of Western Ontario will present:
Discovery of Primary, Cofactor, and Novel Transcription Factor Binding Site Motifs by Recursive, Thresholded Entropy Minimization
by Ruipeng Lu 1, Eliseos Mucaki 2, and Peter Rogan 1,2,3. Departments of (1) Computer Science and (2)Biochemistry, University of Western Ontario, and (3)Cytognomix Inc., London ON
at Compute Ontario (abstract), Conestoga College, Cambridge Ontario, Canada. The presentation is in Room A2107 at 13:55 at the Conestoga College Institute of Technology and Advanced Learning.
May 14, 2015. Presentation at the Great Lakes Chromosome Conference
Dr. Joan Knoll, Chief Scientific Officer of Cytognomix, will present:
Localized, Structural Differences in Condensation of Homologous Metaphase Chromosomes and the Underlying Mechanism
at the 53rd Annual Great Lakes Chromosome Conference at the University of Toronto, Ontario, Canada.
April 30. Sale of MutationForecaster subscription
Cytognomix has sold an annual subscription to MutationForecaster, our comprehensive solution for next generation sequencing based mutation interpretation, to a hospital in Toronto, Ontario Canada. This customer was a previous subscriber to the Automated Site and Exon Definition Server, which is embedded in MutationForecaster and no longer available as standalone software.
April 30, 2015. Sale of Shannon mRNA splicing mutation pipeline software license
Cytognomix’s standalone version of the Shannon mRNA splicing mutation analysis pipeline is distributed through Qiagen CLC bio as a plug-in for their Genomics Workbench and Server software.This product was recently purchased by Dr. Hidetaka Eguchi at the Research Center for Genomic Medicine, Saitama Medical University, Japan.
April 6, 2015. New paper published on cancer of unknown primary
Collaborative effort led by Greg Zaric at the University of Western Ontario:
Identification and survival outcomes of a cohort of patients with cancer of unknown primary in Ontario, Canada. Kim CS1, Hannouf MB, Sarma S, Rodrigues GB, Rogan PK, Mahmud SM, Winquist E, Brackstone M, Zaric GS. Acta Oncol. 2015 Mar 31:1-7. (link)
April 3, 2015. Video tutorials available for MutationForecaster
Videos are now available describing how to use and interpret results from the different components of the MutationForecaster system. Credits: Ben Shirley, Shannon Brown.
Please go to this link to view them all: link
For example, this is a general overview of the system: