Cytognomix has a well-maintained portfolio of multiple issued and pending patents covering its intellectual property which it either owns or licenses:
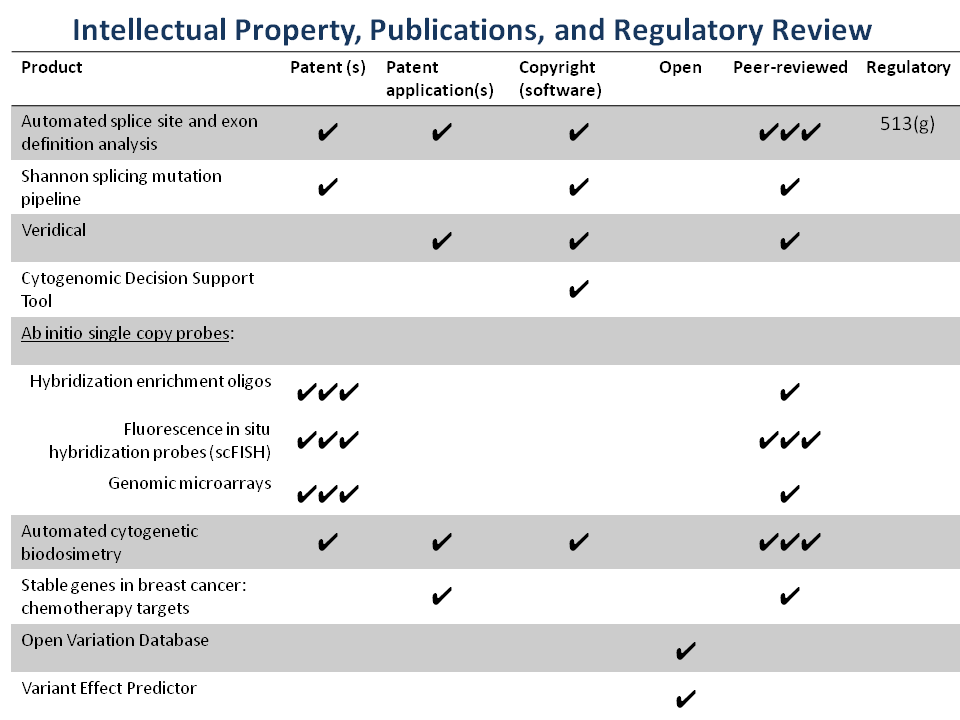
Complete list of Patents and Patent applications
Information theory based binding site analysis. This invention detects and quantifies the strengths of binding sites in nucleic acids. Binding sites are defined based upon the individual information content of a particular site of interest (US Patent 5,867,402). Substitutions within the binding site sequences can be analyzed to determine whether the substitution will cause a deleterious mutation or a benign polymorphism. In addition, new binding sites can be identified using individual information content. Further a computer system is described for determining and displaying individual information content of a binding site sequence. Several software products have been created and distributed based on this technology, including the ASSA server and the Shannon pipeline. ASSA has be upgraded to the ASSEDA server (Mucaki et al., 2013) which extends this technology to predict mutant mRNA isoforms generated from individual mutations. This works by defining the total individual information of a set of binding sites (including splicing regulatory sites) that recognize a common functional unit, which, in this instance, is an exon (US Pat. App. 14/154,905). We have also implemented our Shannon pipeline software which uses this technology on a genome scale to analyze the molecular phenotypes of splicing-related gene variants discovered in exomes, genomes or targeted sequencing. Results from the Shannon pipeline are validated with Veridical, software which uses matched RNA-Seq data to corroborate predictions (US Pat. App. 14/594,109).
Single copy technology. As important as the protection, is the fact that the SC technology does not infringe on any patents controlled any other organization. US Patent No. 7,734,424 covers the foundational “abinitio”TM technology currently used in probe and microarray designs. US Patents 8,209,129 and 8,407,013 cover single copy DNA probes which include divergent repetitive sequences, thus significantly extending the portions of the genome that can be used for such probes beyond traditional single copy sequences. The technology also increases the density of genomic DNA probes for higher resolution genetic analysis beyond what is used in FISH, genomic microarrays for array comparative genomic hybridization, and solution capture hybridization arrays for sequence enrichment in deep sequencing. It is licensed to Cytognomix.
Cytognomix’s SC FISH probes are one of several products that come from applications of “Ab-initio” design algorithms. The method depends on finished genome sequences as the raw template for a recursive algorithm that results in “single copy” sequence information (which are unique in the genome sequence) and corresponding oligonucleotide or FISH products. The algorithm can easily be applied to any completed genome sequence, limited only by the quality of the input sequences and processing time. The output is in the form of “single copy intervals”, generally varying size from hundreds to thousands of nucleotides. The ab-initio method makes use of parallel computing resources to reduce the time required to identify single copy intervals and distinguish them from repeated sequences.
The Ab-initio process confers advantages over purely repeat-masked probes, which we previously developed (US Pats. 6,828,097, 7,014,997 and others). The design criteria permit inclusion of highly divergent interspersed repeated sequences that don’t cross-hybridize to other genomic locations. This opens up access to regions of the genome that are not covered by our previous inventions. If desired, ab-initio probes can optionally exclude segmental duplications and self-chain blocks of low copy sequences capable of cross-hybridizing to undesirable genomic targets. Aside from FISH, genomic microarrays, and NGS capture reagents, ab-initio single copy sequences have been used to identify stable genes in breast tumour genomes, whose products are often targets of chemotherapy agents (US Pat. App. Ser. No. 13/744,459, US Patent 9,624,549). This technology has led to novel algorithms and copyrighted software that accurately predict cellular and patient responses tailored to individual chemotherapy drugs (chemotherapy.cytognomix.com).
Cytogenetic biodosimetry. Our genome coordinate-based FISH products led us to develop image processing algorithms for automated detection of chromosome abnormalities, which have been published as papers and patent applications. We created new methods for ranking and segmenting chromosome images, which has led to new applications in biological radiation biodosimetry. These technologies are covered by US Patents 8,605,981 and 10,929,641, German Patent 11 2011103687, and other patents pending (PCT/US11/59257, US Pat. App. Ser No. 17/137,317). This technology completely automates the interpretation of dicentric chromosomes for individuals exposed to gamma radiation. The corresponding software, the Automated Dicentric Chromosome Identifier and Dose Estimator (or ADCI), has been implemented for desktop/notebook and high performance computing systems. Data from thousands of patient samples can be analyzed in a few hours. We have recently demonstrated that ADCI can quantify partial body radiation exposures, which are typically used in radiation oncology therapy. We will seek regulatory approval for use of ADCI as a medical device for use in monitoring therapy.
Other Patents and Patent Applications:
- Chromosome structural abnormality localization with single copy probes. US Patent #7,014,997
- Ab initio generation of single copy probes. US Patent #8,209,129
- Single copy probes and method of generating same. US Patent #6,828,097
- Mitigation of Cot-1 DNA distortion in nucleic acid hybridization. US Pat. No. 7,833,713
- US Patent Application 09/854,867
- US Patent Application 10/786,970
- US Patent Application 10/676,248
- International Patent Application WO 01/088089
- International Patent Application WO 2004/029283
- International Patent Application WO 2005/094291
- Rapid and comprehensive identification of prokaryotic organisms by metagenomic analysis. U.S. Pat. No. 8,532,934
- Genetic identification and validation of Echinacea species. U.S. Pat. No. 7,811,766
- Rapid and comprehensive identification of prokaryotic organisms. US Pat. No. 8,076,104
- Accurate identification of organisms based on individual information content. U.S. Pat. No. 8,527,207
- Computational analysis of nucleic acid information defines binding sites. US Pat. No. 5,867,402
- Method for rapid identification of prokaryotic and eukaryotic organisms. US Pat. No. 5,849,492