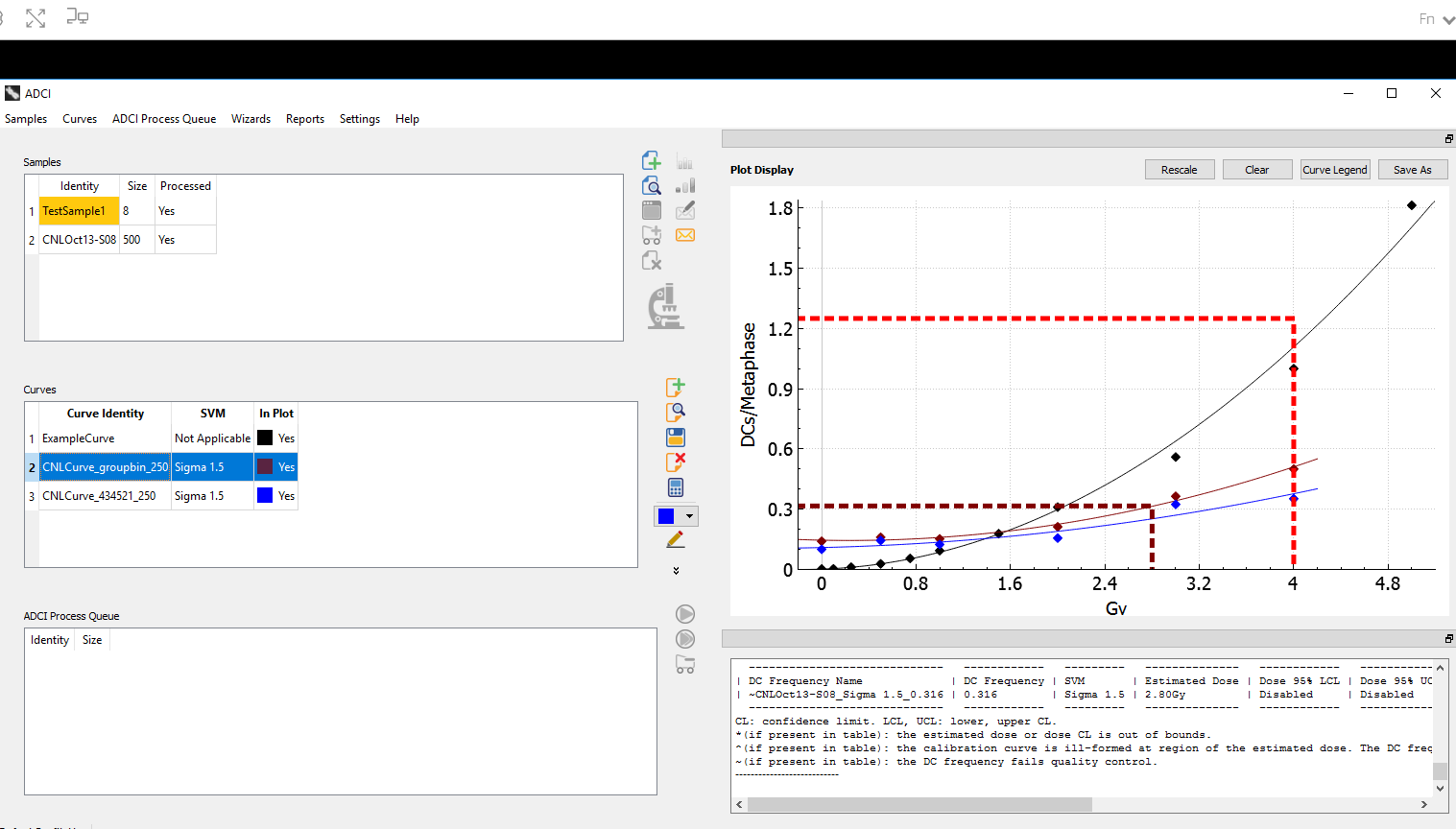
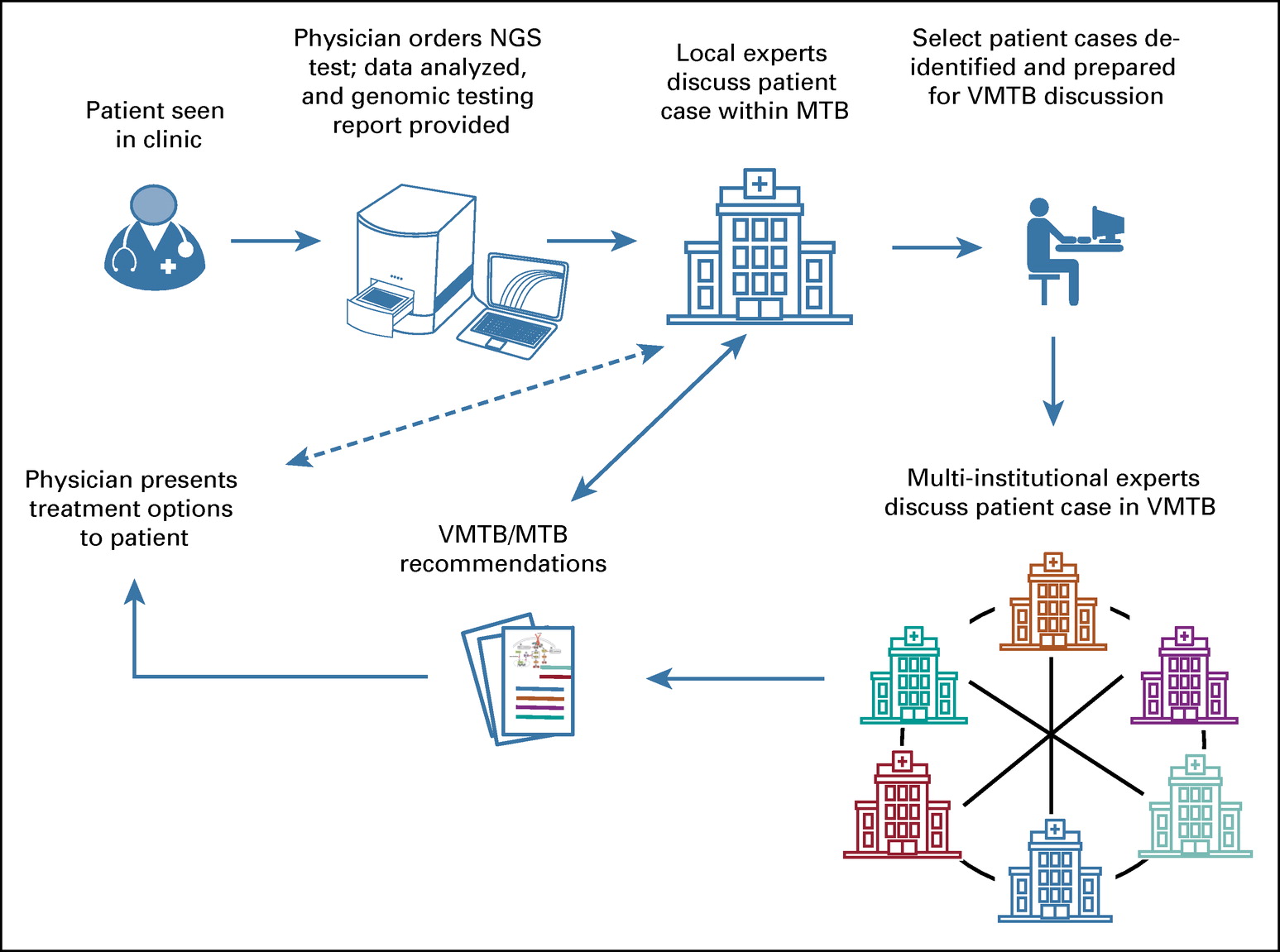
We are giving a platform presentation at the upcoming American Society of Human Genetics virtual meeting #ASHG2020
Session: Personalized Medicine Approaches in Healthcare.
Paper: Pathway-extended gene expression signatures integrate novel biomarkers that improve predictions of responses to kinase inhibitors
P. K. Rogan(1,2), A. J. Bagchee-Clark(1), E. J. Mucaki(1). 1. Department of Biochemistry, University of Western Ontario and 2. CytoGnomix Inc., London ON Canada

Abstract: Individualized chemotherapy selection in cancer potentially maximizes drug efficacy while minimizing drug toxicity. Despite the knowledge of many pharmacogenetic biomarkers, inter-individual variability in response to chemotherapeutic response has limited the success of the approach. We derive multi-gene expression signatures that predict individual patient responses to tyrosine kinase inhibitors (TKIs): erlotinib, gefitinib, sorafenib, sunitinib, lapatinib and imatinib. Gene models for TKIs implicated from the published literature tend to predict either sensitivity or resistance to TKIs well (but not both). This
issue was addressed with a systems biology-based strategy that expanded with candidate gene products related to these genes in these models through biochemical pathways and interactions. Using patient transcriptome data, these Pathway-Extended (PE) models predicted responses for individual patients that matched observed outcomes at accuracies of 65% (imatinib), 71% (lapatinib and gefitinib), 78% (sunitinib), 83% (erlotinib) and 89% (sorafenib). After training and evaluating many extended signatures, those with the strongest predictive performance were composed primarily of pathway-related genes that according to post-hoc analysis were clearly implicated in cancer phenotypes. Machine learning-based PE expression signatures display strong efficacy in predicting both sensitivity and specificity in patients through incorporation
of novel cancer biomarkers.
#genomics #medicine #chemotherapy #cancertreatment #kinaseinhibitors